



Содержание
Перейти к:
https://doi.org/10.21886/2219-8075-2023-14-1-38-42
Перейти к:
Нарушение формирования пола (НФП) — термин, используемый для обозначения врождённых нарушений, повлекших за собой атипичное строение гениталий. Причиной НФП является нарушение эмбрионального развития половой системы вследствие хромосомной, генетической патологии или других неблагоприятных для течения беременности воздействий. НФП влечёт за собой трудности со стороны социальной адаптации семьи, приводит к тяжёлым психологическим расстройствам у ребенка и его родственников. Паспортный пол ребенка с НФП должен быть установлен только после полноценного обследования и консультаций специалистов в данной области. Представлен клинический случай с целью иллюстрации сложности дифференциальной диагностики и выбора паспортного пола у ребенка с одной из редких форм НФП.
Шайдуллина М.Р., Акрамов Н.Р., Валеева Ф.В., Алиметова З.Р., Колбасина Е.В. Клинический случай нарушения формирования пола при кариотипе 47 XYY. Медицинский вестник Юга России. 2023;14(1):38-42. https://doi.org/10.21886/2219-8075-2023-14-1-38-42
Shaydullina M.R., Akramov N.R., Valeeva F.V., Alimetova Z.R., Kolbasina E.V. Clinical сase of disorder of sex development with karyotype 47XYY. Medical Herald of the South of Russia. 2023;14(1):38-42. (In Russ.) https://doi.org/10.21886/2219-8075-2023-14-1-38-42
Термин «нарушение формирования пола» (НФП) в настоящее время считается наиболее уместным для описания заболеваний, характеризующихся несоответствием строения наружных и внутренних половых органов и генетического пола [1]. При выявлении такого состояния у ребенка в клинической практике дальнейшая тактика врачей зависит от полноценного, достоверного и своевременного обследования. Выбор пола в данном случае зависит от множества критериев (результатов генетического, гормонального, ультразвукового и других исследований), а также от способности пациента к дальнейшей репродуктивной функции, возможности адекватного медицинского вмешательства и дальнейшей социализации личности. НФП ребенка является важной психологической проблемой для родителей, которые также должны принимать участие в выборе пола.
Нарушения половой дифференцировки условно делят на три большие группы: НФП 46,XY, НФП 46,XX и сексхромосомные нарушения, при которых регистрируются различные варианты аномального кариотипа.
Впервые синдром Якобса (мужчины 47XYY) описан в 1960 г. [2] и в настоящий момент не считается редкой патологией: согласно различным источникам, кариотип 47XYY регистрируется с частотой от 1 на 1000 [3] до 1 на 10 000 мужчин [4]. Однако до 85% пациентов так и остаются не выявленными [2].
Данная анеуплодия ассоциируется с такими фенотипическими особенностями, как высокорослость, макроцефалия, макроорхидизм, гипертелоризм, клинодактилия [2]. У пациентов с кариотипом 47XYY описана задержка темпов моторного развития на фоне мышечной гипотонии и тремора конечностей [2][3]. Однако чаще всего больные с синдромом Якобса привлекают к себе внимание нарушением нейрокогнитивных функций (дислексией, дисграфией, синдромом гиперактивности, различными расстройствами аутистического спектра и нарушением поведенческих реакций, приводящих к социальной дезадаптации пациентов) [2][3][5]. Несмотря на наличие единичных описаний аномалий развития урогенитального тракта в виде микрофаллуса, крипторхизма, гипоспадии, а также задержки полового развития у мальчиков с кариотипом 47XYY [2], исследования взрослой популяции пациентов свидетельствуют о нормальном или высоком уровне тестостерона [6]. Чаще всего данная аномалия диагностируется у мужчин при обследовании по поводу бесплодия [7] или проведении «сплошного» кариотипирования в тюрьмах и психиатрических клиниках [2][5].
Представленный клинический случай является редким примером нарушения формирования пола, сопряжённого с кариотипом 47 XYY, и отражает сложности выбора паспортного пола у детей с патологией строения половых органов.
Ребенок С. родился от третьей беременности, протекавшей на фоне ОРЗ в первом триместре, угрозы прерывания беременности в 29 недель, вторых срочных родов. Рост при рождении — 54 см, масса — 3450 г, 7–8 баллов по шкале Апгар. В связи с неправильным строением наружных гениталий переведён в отделение патологии новорожденных, затем в эндокринологическое отделение для дальнейшего обследования с диагнозом «Нарушение формирования пола. Неонатальная желтуха. Внутриутробная инфекция неуточненной этиологии. Инфекция мочевыводящих путей. Перинатальное поражение центральной нервной системы. Малые аномалии развития сердца (открытое овальное окно)».
В первый день жизни проведено исследование уровня 17-ОН-прогестерона (6,0 нмоль/л, при норме (N) до 15 нмоль/л) для исключения жизнеугрожающего состояния — сольтеряющей формы врожденной дисфункции коры надпочечников. По данным ультразвукового исследования (УЗИ), в возрасте 1 недели правое яичко визуализируется в проекции средней трети пахового канала, левое достоверно не определяется, за мочевым пузырем обнаружена матка. Кариотипирование в возрасте 1 недели выявило хромосомную патологию — 47, XYY. Повторное определение кариотипа в возрасте 2 недель дало следующий результат — мозаицизм по половым хромосомам 47, XYY (142)/45, X0 (8).
Проведённая в возрасте 1 месяца оценка эндокринного статуса подтвердила присутствие тестикулярной ткани у ребенка: базальные уровни тестостерона, антимюллерова гормона (АМГ), ингибина В (табл. 1), положительная проба с хорионическим гонадотропином (тестостерон исходно — 6,94 нмоль/л, через 24 часа после инъекции № 3 по 250 МЕ — 13,87 нмоль/л). В связи с этим было поддержано решение родителей о выборе мужского паспортного пола.
Таблица / Table 1
Результаты обследования ребенка в возрасте 1 и 16 месяцев
The results of examination of the child at the age of 1 and 16 months
|
Параметр (единицы измерения) Unit parameter |
Уровень в возрасте 1 месяца Level at 1 month of age |
Уровень в возрасте 16 месяцев Level at 16 month of age |
Референсный интервал Reference interval |
|
ФСГ (мМе/мл) FSH (mIU/ml) |
7,54 |
5,8 |
муж. 0–1 год <3,5 жен. 0–1 год 1,84–20,26 men 0–1 year <3,5 women 0–1 year 1,84–20,26 |
|
ЛГ (мМе/мл) LH (mIU/ml) |
3,6 |
1,1 |
0–1 год <6,34 0–1 year <6,34 |
|
Тестостерон (нмоль/л) Testosterone (nmol/l) |
6,1 |
˂0,025 |
муж. 4 дня–6 мес. 0,30–10,36 жен. 4 дня – 9 лет <2,15 men 4 days–6 months 0,30–10,36 women 4 days–9 years <2,15 |
|
Эстрадиол (пг/мл) Estradiol (pg/ml) |
|
˂5 |
муж. 0–1 год <86 жен. 0–1 год <155 men 0–1 year <86 women 0–1 year <155 |
|
Антимюллеров гормон (нг/мл) AMH (ng/ml) |
16,91 |
0,8 |
муж. препубертат: 3,80–159,80 жен. препубертат <8.90 pre–pubertal men 3,80–159,80 pre–pubertal women <8.90 |
|
Ингибин B (пг/мл) Inhibin B (pg/ml) |
65,95 |
3,2 |
муж. 0–18 лет 4,0–352,0 жен. 0–18 лет <83,0 men 0–18 years 4,0–352,0 women 0–18 years <83,0 |
В возрасте 1 года и 4 месяцев ребенок вновь поступает в отделение эндокринологии детской многопрофильной больницы для подготовки к маскулинизирующей пластике гениталий.
При объективном осмотре у мальчика констатировано неправильное строение наружных гениталий: гипоспадия, мошоночная форма; расщепленная мошонка; двусторонний крипторхизм, микрофаллус (2,0 см). Наружное отверстие мочеиспускательного канала открывается у основания полового члена между частями расщепленной мошонки (рис.1). Гонады в мошонке не пальпируются.
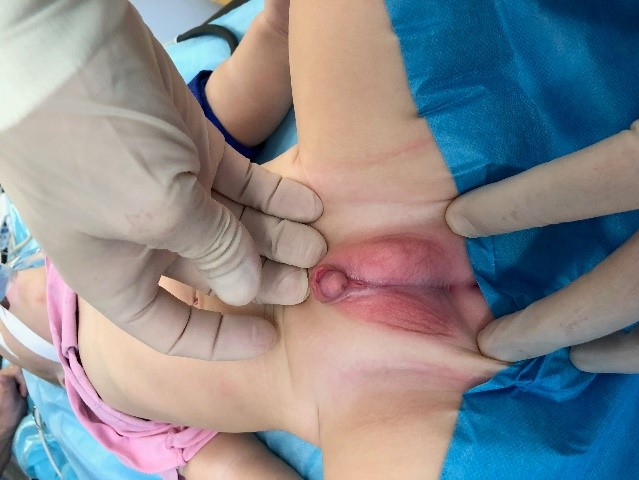
Рисунок 1а. Внешний вид наружных половых органов
Figure 1a. The appearance of the external genitalia

Рисунок 1б. Гипоспадия, мошоночная форма
Figure 1b. Scrotal hypospadias
Исследование уровня гормонов свидетельствовало о значимом динамическом снижении не только уровня тестостерона, что естественно при выходе из возраста минипубертата, но АМГ и Ингибина В.
Ребенку проведено повторное исследование кариотипа методом FISH, установлено отсутствие мозаицизма, подтверждено наличие дисомии по Y-хромосоме (47, XYY — 100%). В связи с предположением о полном отсутствии или нарушениях в структуре гена SRY (Sex-determining Region Y) проведена оценка последовательности Y-хромосомы SRY, патогенных и вероятно патогенных вариантов мутаций гена SRY не обнаружено. Проведена молекулярно-генетическая диагностика (панель «Нарушение формирования пола» 38 генов) патогенных и вероятно патогенных вариантов, а также вариантов с неизвестной клинической значимостью, объясняющих причину заболевания на молекулярно-генетическом уровне, не обнаружено.
С целью определения плана оперативного вмешательства проведена панэндокопия (цистоуретроскопия, кольпоскопия): между наружным и внутренним сфинктерами урогенитального тракта обнаружен вход во влагалище, влагалище длиной 6 см (рис. 2).
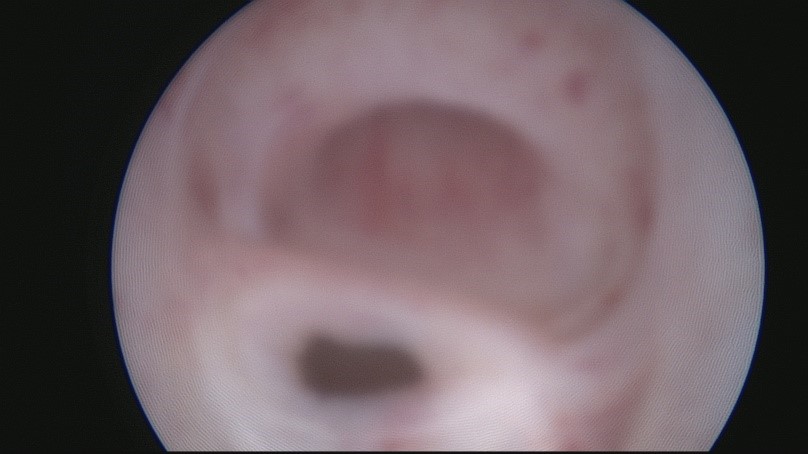
Рисунок 2. Цистоскопия
Figure 2. Сystiscopy
При лапароскопической ревизии брюшной полости обнаружено следующее: левая гонада округлой формы, придаток гиперемирован, плотный. Правая гонада на конце маточной трубы, дисплазирована. Внешний вид обеих гонад не типичен ни для яичка, ни для яичника (рис.3а, 3б). Левое паховое кольцо не облитерировано. Имеется матка маленьких размеров.

Рисунок 3а. Внешний вид дисплазированных гонад: левая гонада диаметром 1 см
Figure 3а. Appearance of dysplastic gonads: the left gonad with a diameter of 1 cm
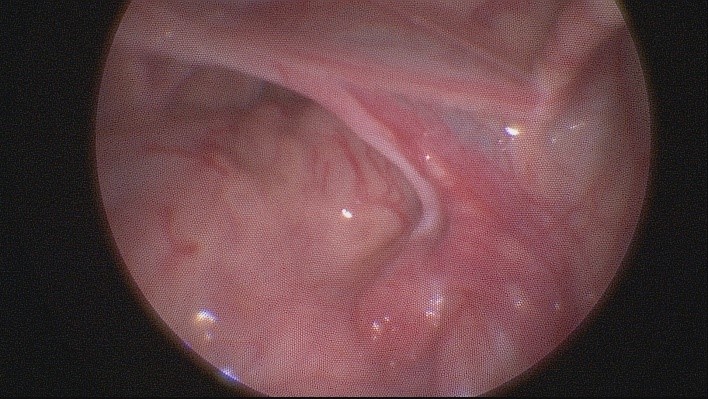
Рисунок 3б. Внешний вид дисплазированных гонад: правая гонада
Figure 3b. Appearance of dysplastic gonads: the right gonad
Консилиумом специалистов, принимавших участие в обследовании ребенка, установлен диагноз «Нарушение формирования пола; сексхромосомные аномалии 47, XYY; дисгенезия гонад». В связи с выставлением абсолютных показаний к гонадэктомии (дисгенезия, расположение гонад в брюшной полости связано с высоким риском малигнизации [8]), наличием матки, влагалища и возможностью проведения одноэтапной феминизирующей пластики гениталий пациенту рекомендована смена паспортного пола на женский. В случае решения семьи о выборе мужского паспортного пола рекомендованы гонадэктомия и многоэтапная маскулинизирующая пластика наружных половых органов.
Несмотря на предупреждения о возможных рисках и осложнениях от предстоящих медицинских вмешательств, рекомендации психолога и других специалистов, родители приняли решение о сохранении мужского пола ребенку.
Анализ литературных данных (достаточная маскулинизация наружных гениталий при рождении у подавляющего большинства мальчиков с кариотипом 47, XYY), отсутствие выявленных аномалий Y-хромосомы и мутаций, объясняющих причину заболевания у данного пациента, не позволяет исключить возможность тератогенного воздействия, приведшего к формированию дизгенезии гонад на ранних сроках беременности, недостаточной внутриутробной секреции АМГ (наличие матки, фаллопиевых труб) и тестостерона, что и привело к неправильному формированию пола у ребенка.
1. Hughes IA, Houk C, Ahmed SF, Lee PA; Lawson Wilkins Pediatric Endocrine Society/European Society for Paediatric Endocrinology Consensus Group. Consensus statement on management of intersex disorders. J Pediatr Urol. 2006;2(3):148-62. DOI: 10.1016/j.jpurol.2006.03.004
2. Bloy L, Ku M, Edgar JC, Miller JS, Blaskey L, Ross J, Roberts TPL. Auditory evoked response delays in children with 47,XYY syndrome. Neuroreport. 2019;30(7):504-509. DOI: 10.1097/WNR.0000000000001233
3. Stochholm K, Juul S, Gravholt CH. Socio-economic factors affect mortality in 47,XYY syndrome-A comparison with the background population and Klinefelter syndrome. Am J Med Genet A. 2012;158A(10):2421-9. DOI: 10.1002/ajmg.a.35539
4. Berglund A, Viuff MH, Skakkebæk A, Chang S, Stochholm K, Gravholt CH. Changes in the cohort composition of turner syndrome and severe non-diagnosis of Klinefelter, 47,XXX and 47,XYY syndrome: a nationwide cohort study. Orphanet J Rare Dis. 2019;14(1):16. DOI: 10.1186/s13023-018-0976-2
5. Green T, Flash S, Reiss AL. Sex differences in psychiatric disorders: what we can learn from sex chromosome aneuploidies. Neuropsychopharmacology. 2019;44(1):9-21. DOI: 10.1038/s41386-018-0153-2
6. Ross JL, Roeltgen DP, Kushner H, Zinn AR, Reiss A, et al. Behavioral and social phenotypes in boys with 47,XYY syndrome or 47,XXY Klinefelter syndrome. Pediatrics. 2012;129(4):769-78. DOI: 10.1542/peds.2011-0719
7. Borjian Boroujeni P, Sabbaghian M, Vosough Dizaji A, Zarei Moradi S, Almadani N, et al. Clinical aspects of infertile 47,XYY patients: a retrospective study. Hum Fertil (Camb). 2019;22(2):88-93. DOI: 10.1080/14647273.2017.1353143
8. Fallat ME, Donahoe PK. Intersex genetic anomalies with malignant potential. Curr Opin Pediatr. 2006;18(3):305-11. DOI: 10.1097/01.mop.0000193316.60580.d7
Шайдуллина Мария Рустемовна, к.м.н., доцент кафедры эндокринологии; заведующий врач-детский эндокринолог
Казань
Авторы заявляют об отсутствии конфликта интересов.
Акрамов Наиль Рамилович, д.м.н., профессор кафедры детской хирургии
Казань
Авторы заявляют об отсутствии конфликта интересов.
Валеева Фарида Вадутовна, д.м.н., профессор, заведующая кафедрой эндокринологии
Казань
Авторы заявляют об отсутствии конфликта интересов.
Алиметова Зульфия Раисовна, к.м.н, ассистент кафедры эндокринологии
Казань
Авторы заявляют об отсутствии конфликта интересов.
Колбасина Елена Владимировна, заведующая отделением эндокринологии
Новгород
Авторы заявляют об отсутствии конфликта интересов.
Шайдуллина М.Р., Акрамов Н.Р., Валеева Ф.В., Алиметова З.Р., Колбасина Е.В. Клинический случай нарушения формирования пола при кариотипе 47 XYY. Медицинский вестник Юга России. 2023;14(1):38-42. https://doi.org/10.21886/2219-8075-2023-14-1-38-42
Shaydullina M.R., Akramov N.R., Valeeva F.V., Alimetova Z.R., Kolbasina E.V. Clinical сase of disorder of sex development with karyotype 47XYY. Medical Herald of the South of Russia. 2023;14(1):38-42. (In Russ.) https://doi.org/10.21886/2219-8075-2023-14-1-38-42
344022, г. Ростов-на-Дону, пер. Нахичеванский, 29
Ростовский государственный медицинский университет
Тел.: +7 918 571 0558
E-mail: journal@medicalherald.ru


































