


Содержание
Перейти к:
 А. А. Лебеденко,
С. Б. Бережанская,
А. С. Тодорова,
Н. Н. Вострых,
Е. Я. Каушанская,
Е. А. Лукьянова,
Е. А. Папшева,
Г. Н. Смыкова,
Л. Н. Тараненко
А. А. Лебеденко,
С. Б. Бережанская,
А. С. Тодорова,
Н. Н. Вострых,
Е. Я. Каушанская,
Е. А. Лукьянова,
Е. А. Папшева,
Г. Н. Смыкова,
Л. Н. Тараненко https://doi.org/10.21886/2219-8075-2020-11-4-84-91
Перейти к:
Глутаровая ацидурия I типа (недостаточность глутарил-КоА-дегидрогеназы, глутаровая ацидемия I типа) – редкое аутосомно-рецессивное заболевание, обусловленное мутациями в гене, кодирующем фермент глутарил-КоАдегидрогеназа (GCDH). Церебральную органическую ацидурию, вызванную дефицитом глутарил-КоА-дегидрогеназы, принято, в первую очередь, считать неврологическим расстройством.
Фенотипический спектр нелеченной GA-1 варьирует от более распространенной и выраженной формы (болезнь с младенческим началом) до малосимптомной и менее распространенной формы. У людей с одним и тем же генотипом клинические проявления и глубина поражения ЦНС могут широко варьировать в зависимости от возраста манифестации острых энцефалопатических кризов. Предполагается, что при раннем выявлении и начале лечения «бессимптомных” новорожденных (в условиях скрининга на указанное заболевание) большинство людей, у которых развились бы проявления GA-1 с детским или поздним началом, останутся бессимптомными.
Лебеденко А.А., Бережанская С.Б., Тодорова А.С., Вострых Н.Н., Каушанская Е.Я., Лукьянова Е.А., Папшева Е.А., Смыкова Г.Н., Тараненко Л.Н. Редкий случай глутаровой ацидурии I типа у ребенка раннего возраста. Медицинский вестник Юга России. 2020;11(4):84-91. https://doi.org/10.21886/2219-8075-2020-11-4-84-91
Lebedenko A.A., Berezhanskay S.B., Todorova A.S., Vostrykh N.N., Kaushanskay E.Y., Lukyanova E.A., Papsheva E.A., Smykova G.N., Taranenko L.N. A rare case of type i glutaric aciduria in an early child. Medical Herald of the South of Russia. 2020;11(4):84-91. (In Russ.) https://doi.org/10.21886/2219-8075-2020-11-4-84-91
Глутаровая ацидурия I типа (ГА-1) на основании постановления Правительства Российской Федерации от 2012 г. внесена в регистр жизнеугрожающих и хронических, прогрессирующих редких (орфанных) заболеваний, приводящих к сокращению продолжительности жизни или инвалидности.
Органическая ацидурия относится к наследственным болезням обмена веществ, занимающим важное место в педиатрической неврологии, и характеризуется повышенной экскрецией органических кислот с мочой. Глутаровая ацидурия I типа (недостаточность глутарилКоА-дегидрогеназы, глутаровая ацидемия I типа) — аутосомно-рецессивное заболевание, обусловленное мутациями в гене, кодирующем фермент глутарил-КоА-дегидрогеназа (GCDH).
Церебральную органическую ацидурию, вызванную дефицитом глутарил-КоА-дегидрогеназы, принято считать неврологическим расстройством. Однако некоторые публикации оспаривают эту точку зрения, демонстрируя, что периферическая нервная система [1] и почки также могут быть вовлечены в длительное течение заболевания [2]. GCDH ген сопоставлен с хромосомой 19p13.2 и кодирует флавин-адениндинуклеотид-зависимый белок митохондриального матрикса, который участвует в деградации L-лизина, L-гидроксилизина и L-триптофана. На сегодняшний день опубликовано 187 (подтвержденных или вероятных) патогенных мутаций, перечисленных в базе данных мутаций генов человека (данные от 18 апреля 2016 г.). Наиболее распространенной является Аrg402 Trp. В европейских странах частота встречаемости указанной мутации составляет 12 – 40 % [3][4].
Биохимически GA-I характеризуется накоплением глутаровой кислоты (GA), 3-гидроксиглутаровой кислоты (3-OH-GA), глутаконовой кислоты и глутарилкарнитина (C5DC). Эти метаболиты могут быть обнаружены в жидкостях организма (моча, плазма, спинномозговая жидкость) и тканях с помощью газовой хроматографии/масс-спектрометрии (GC/MS) или тандемной массспектрометрии с электрораспылительной ионизацией. Возможна пренатальная диагностика на основании определения концентрации глутаровой кислоты в амниотической жидкости или активности GCDH в культуре амниотических клеток, а также путем мутационного анализа гена GCDH ДНК хорионических ворсин. В ряде стран с конца 1990-х гг. проводится скрининг новорожденных, поскольку раннее выявление заболевания существенно снижает частоту манифестации или тяжесть и выраженность клинических проявлений и морфологических изменений в головном мозге, хотя показатели смертности и МРТ- изменений в головном мозге у больных, взятых под наблюдение и получавших терапию после неонатального скрининга до конца не изучены [5].
Это редкое наследственное заболевание обмена веществ впервые описано Goodman et al. в 1975 г., после чего в мире было зарегистрировано более 500 случаев [6]. Распространенность во всем мире составляет от 1:30000 до 1:110000 [7].
Фермент GCDH, локализованный в матриксе митохондрий, участвует в метаболизме аминокислот лизина, гидролизина, триптофана и катализирует две последовательные реакции превращения глутарил-КоА в кротонил-КоА, в результате чего и происходит накопление глутаровой и 3-OH-глутаровой кислот в биологических жидкостях и тканях, которые оказывают нейротоксическое действие преимущественно на подкорковые структуры головного мозга. Таким образом, основную роль в патофизиологии заболевания играют эксайтотоксические последствия, вызванные глутаровой и 3-гидроксиглутаровой кислотами, обладающими сходной структурой с аминокислотой глутаматом [3][8].
Изначально ГА-1 была произвольно разделена на два биохимических подтипа: высокий уровень экскреции мочевой глутаровой кислоты (ГК) и низкий уровень экскреции мочевой ГК. Второй тип сопровождается сохранением до 30 % остаточной глутарил-КоА-дегидрогеназной активности [5]. Однако у детей не выявлено четкой корреляции биохимических подтипов с клиническим фенотипом заболевания. Предполагается, что независимо от уровня экскреции мочевой ГК больные дети подвержены одинаковому риску поражения подкорковых структур [9]. В то же время в динамике заболевания выявляют повышенную распространенность прогрессирующих с возрастом поражений белого и серого вещества при МРТ [10] и повышенные внутримозговые концентрации GA и 3-OH-GA, обнаруженные in vivo с помощью протонной магнитно-резонансной спектроскопии [1 H-MRS] при высокой экскреции метаболита GA с мочой [11].
У детей по клиническому течению выделяют два основных подтипа ГА-1 — с острым «энцефалитоподобным» и подострым (доброкачественным) течением. В течение первых месяцев дети могут развиваться относительно нормально, в неврологическом статусе отмечаются раздражительность, повышенная нервно-рефлекторная возбудимость, легкая гипотония. Наиболее ранним, но не строго специфичным признаком является макроцефалия с рождения или макроцефалия, появляющаяся в течение первых месяцев жизни. Острая неврологическая симптоматика, напоминающая энцефалит, развивается в течение первых двух лет жизни, но чаще на первом году и характеризуется возникновением критических состояний-метаболических «энцефалитоподобных» кризов, начинающихся срыгиваниями, рвотой, кишечными расстройствами, лихорадкой, судорожными приступами, угнетением сознания вплоть до сопора и комы, в результате развивающегося отека и набухания мозга, иногда приводящего к летальному исходу в первые сутки заболевания [12].
Далее на фоне сохраняющегося метаболического ацидоза сохраняются сонливость, гипотония, ригидность, дистония, эпилептические приступы. В результате перенесенных кризов могут развиваться некрозы базальных ганглиев и стойкий неврологический дефицит. Через несколько дней или недель после криза появляются различные типы гиперкинезов (орофациальные, хореиформные, хореоатетоидные, баллистические), усиливается односторонняя или диффузная мышечная дистония, часто сочетающаяся со спастичностью. После кризов восстановление неврологических нарушений происходит медленно и частично, с формированием вторичных осложненияй (хронический аспирационный синдром, подвывихи суставов, болевой синдром). Далее заболевание приобретает волнообразный характер [13,][14].
В 25 % случаев у больных при отсутствии «энцефалитоподобных» кризов возможна задержка психомоторного развития с постепенной утратой ранее приобретенных навыков и присоединением различных видов гиперкинезов, что характеризует более доброкачественное течение заболевания, но дистонические гиперкинезы могут приводить к нарушению важных функций (двигательных, речевых, мелкой моторики (письма). В целом, картина укладывается в спастико-гиперкинетическую форму детского церебрального паралича, и именно с этим диагнозом многие дети длительное время наблюдаются у неврологов. Отмечаются частые эпизоды немотивированной лихорадки, эпилептические приступы. Возможны офтальмологические нарушения, которые имеют многообразные проявления, характеризующиеся кровоизлияниями в сетчатку, катарактой, офтальмопарезом, косоглазием и пигментной дегенерацией сетчатки. У нелеченных больных с ранним дебютом заболевания смертельный исход может наступить в течение первого десятилетия жизни на фоне тяжелого метаболического криза или в результате развития Рейе-подобного синдрома.
После появления неврологических симптомов клиническая картина острой и хронической форм заболевания становится сходной. В период интеркуррентных инфекций или травм течение может сопровождаться острыми эпизодами комы, судорожных приступов, кетоза, гипогликемии. Риск энцефалопатических кризов превалирует до трех лет. Профилактическое лечение приблизительно в 75 % случаев может приводит к относительно нормальному развитию, в то же время существует категория больных с высоким риском прогрессирования заболевания, даже несмотря на своевременно начатое лечение.
На сериях МРТ и КТ у пациентов, наблюдаемых с рождения, отмечаются прогрессирующие поражения
центральной нервной системы, которые могут предшествовать клинической стадии заболевания. Наиболее характерными проявлениями являются расширение сильвиевой борозды, поражение цистерн среднего мозга, острые субдуральные гематомы или хронические субдуральные скопления, гипоплазия височных долей. У пациентов с острой энцефалоподобной формой заболевания более часто выявляется выраженная лобно-височная атрофия, которая сочетается с диффузной атрофией коры и областями глубоких аномалий развития белого вещества головного мозга. Нередко регистрируется изолированное поражение скорлупы или в совокупности с поражением хвостатого ядра [15].
Механизмы развития ГА-1 до конца не изучены. Преимущественное поражение стриарной системы связывают с избирательной токсичностью глутаровой кислоты и / или её производных. Также глутаровая кислота и ее метаболиты ингибируют декарбоксилазу глутаминовой кислоты (фермент, участвующий в метаболизме ГАМК), что вызывает снижение концентрации этого тормозного нейромедиатора. Радиологическими исследованиями выявлено значительное снижение декарбоксилазной активности в лобных отделах коры головного мозга, хвостатых ядрах и скорлупе, определено снижение концентрации ГАМК в цереброспинальной жидкости у больных с ГА-1. Не исключено, что данные биохимические нарушения могут быть вторичными и возникать вследствие повреждений ГАМК-эргических нейронов мозга [16].
Механизмы патогенеза острых «энцефалитоподобных» кризов также окончательно неясны. Считают, что глутаровая и 3-OH-глутаровая кислоты, имеющие структурное сходство с глутаматом, взаимодействуют сN-метил аспартатными рецепторами, для которых глутамат является естественным активатором, что вызывает чрезмерное накопление ионов Са2+ в постсинаптических нейронах и приводит к гибели клеток. Другим возможным нейротоксическим фактором считают накопление промежуточного метаболита обмена триптофана и лизина — квинолиновой кислоты. В настоящее время до конца не распознана причина лобно-височной атрофии и / или гипоплазии и субдуральных кровоизлияний при ГА-1. Высказано предположение, что во время эмбриогенеза 3-ОН-глутаровая кислота может нарушать формирование стенок сосудов, в первую очередь, сосудов головного мозга, приводя к повышению их проницаемости и возникновению кровоизлияний [8][12].
При аутопсии у всех пациентов с ГА-1 выявляют выраженную субкортикальную и кортикальную атрофии головного мозга, атрофическую вентрикуломегалию. В большинстве случаев визуализируются некротические изменения в области скорлупы и головки хвостатого ядра. Реже поражаются зрительные тракты, мозолистое тело, внутренняя капсула, глубокие отделы белого вещества мозжечка, ствола мозга. У ряда пациентов обнаруживают «губчатую» дегенерацию белого вещества, преимущественно в перивентрикулярных областях, реже в таламусе, бледном шаре и стволе головного мозга [8][12].
В связи с редкостью и многообразием, тяжестью и неоднозначностью клинического течения глутаровой ацидурии I типа приводим собственное наблюдение.
Ребенок Вячеслав Н. родился 22.06.2016 г., от третьей беременности (первая — роды, ребенок здоров; вторая — неразвивающаяся), протекавшей на фоне персистенции герпес-вирусной инфекции, токсикоза 2 половины, вторых родов в срок, оперативным путем с обвитием пуповины вокруг шеи массой 4000 г, длиной 53 см с оценкой по шкале Апгар 7 – 8 баллов.
На первом месяце жизни у ребенка отмечалась резкая прибавка окружности головы (+ 5 см). В возрасте 2 месяцев, по данным НСГ, выявлены кисты в височных отделах головного мозга и признаки расширения ликворных
пространств. Мать ребенка обратилась за консультацией к неврологу в частный центр с жалобами на сниженный аппетит, вялое сосание, низкую прибавку массы, сниженные мышечный тонус и двигательную активность. На основании жалоб, результатов нейросонографии, данных объективного осмотра по месту жительства был поставлен диагноз: Перинатальное гипоксически-ишемическое поражение ЦНС, гипертензионно-гидроцефальный синдром. В ходе назначенного лечения, включавшего ноотропные препараты, курсы массажа, ФТЛ, Бобат-терапию, ребенок, со слов матери, постепенно приобрел моторные навыки. Однако, в возрасте 5 месяцев на фоне респираторной инфекции (острый бронхит) развился первый метаболический криз в виде закатывания, кратковременного нарушения дыхания, клонико-тонических судорог, после чего отмечалась утрата приобретенных моторных навыков, появилась тотальная мышечная гипотония, кратковременные миоклонии. В возрасте 6 – 8 месяцев ребенок получил повторно реабилитационную терапию без особого эффекта. В 9 – 10 месяцев появились гиперкинезы рук, асимметрия лица, опущение правого угла рта. К 1 году сформировалась спастичность верхних и нижних конечностей.
В возрасте 1 года 3 месяцев мальчик впервые поступил в педиатрическое отделение № 2 ФГБОУ ВО РостГМУ с диагнозом: Резидуально-органическое поражение ЦНС, спастический тетрапарез, задержка стато-моторного (самостоятельно не сидел, не стоял) и психо-речевого развития (произносил звуки, слоги, понимал речь матери на бытовом уровне). Ребенок был консультирован врачомгенетиком, и с целью исключения наследственного заболевания проведена тандемная масс-спектрометрия, по результатам которой выявлено снижение концентрации свободного карнитина и резкое повышение концентрации глутарилкарнитина, что позволило предположить вероятность глутаровой ацидурии I типа. При определении органических кислот в моче выявлен высокий уровень глутаровой кислоты -1645 мМ/М креатинина. Данные изменения свидетельствовали в пользу предполагаемого диагноза. Одновременно проведена МРТ головного мозга, выявившая МР-картину перивентрикулярных глиозных изменений, дилатации наружных и внутренних ликворосодержащих пространств головного мозга с явлениями субатрофии корковой поверхности лобно-височно-теменных долей полушарий головного мозга, преимущественно височных долей (рис. 1). По данным 12-часового видео-ЭЭГ-мониторинга эпилептических приступов, паттернов эпилептических приступов и эпилептиформной активности в ходе исследования не зарегистрировано. Для уточнения диагноза генетиком рекомендовано проведение частичного анализа гена GCDH методом прямого автоматического секвенирования.
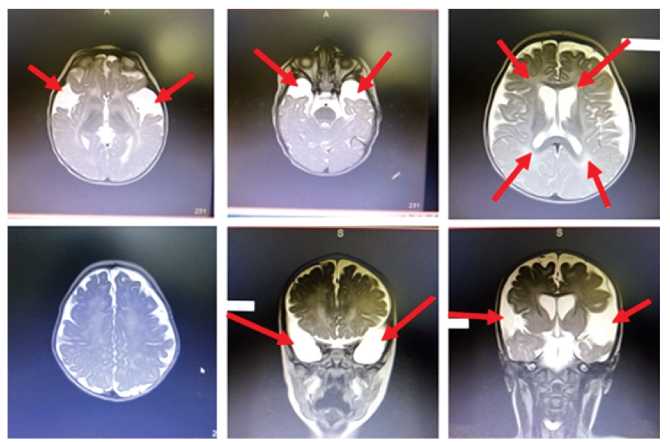
В режиме Т2ВИ определяется выраженное расширение субарахноидальных пространств головного мозга, субатрофия коры головного мозга (преимущественно височных долей), повышение МР сигнала в перивентрикулярном белом веществе головного мозга.
In the T2VI mode, there is a pronounced expansion of the subarachnoid spaces of the brain, subatrophy of the cerebral cortex (mainly the temporal lobes), and an increase in the Mr signal in the periventricular white matter of the brain.
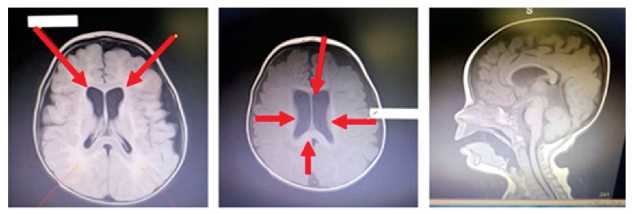
В режиме TIRM (подавление жира) определяется повышение МР сигнала в перивентрикулярном белом веществе головного мозга.
In TIRM mode (fat suppression) an increase in the Mr signal in the periventricular white matter of the brain is determined.
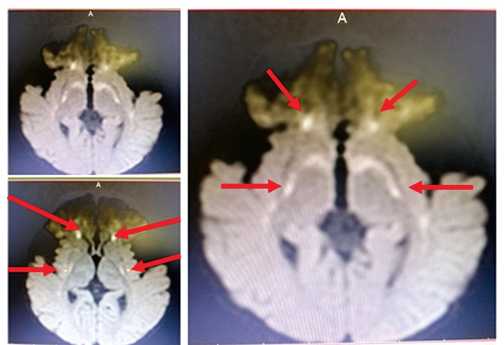
В режимах ДВИ (диффузионно взвешенных при b = 1000) определяется повышение МР сигнала в кортикоспинальных трактах головного мозга
In DVI modes (diffusionally weighted at b =1000), an increase in the MR signal in the corticospinal tracts of the brain is determined
Рисунок 1. МРТ головного мозга ребенка с глутаровой ацидурией I типа.
Figure 1. MRI of the brain of a child with glutaric aciduria of the I type.
Ребенок был направлен в отделение медицинской генетики ФГБОУ РДКБ имени Н.И. Пирогова в возрасте 1 года 7 месяцев для обследования с целью уточнения диагноза и дальнейшего лечения. При проведении частичного анализа гена GCDH методом прямого автоматического секвенирования в 10 экзоне выявлена мутация с.1204С > Т (р.Аrg402Trp) в гетерозиготном состоянии, что позволило установить окончательный диагноз — глутаровая ацидурия I тип. Далее ребенок неоднократно находился в отделении медицинской генетики ФГБОУ РДКБ с диагнозом: Глутаровая ацидурия I тип. Синдром детского церебрального паралича, спастико-гиперкинетический синдром. Задержка психоречевого развития (таблица 1).
Таблица / Table 1
Динамика показателей органических кислот в крови и моче обследованного ребенка (2018 – 2019)
Dynamics of indicators of organic acids in the blood and urine of the examined child (2018 – 2019)
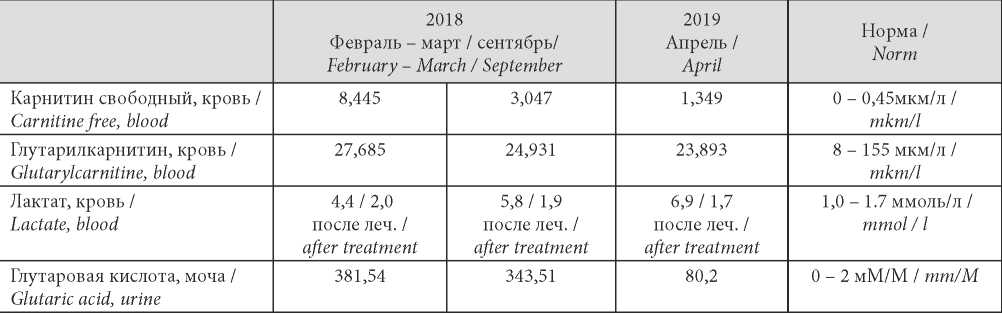
За период пребывания в отделении медицинской генетики ФГБОУ РДКБ комплекс обследования включал консультации диетолога (рекомендована низкобелковая диета из расчета 1,5 г белка на килограмм массы в сутки,дополнительное питание в виде фруктового сока и пюре), офтальмолога (ангиопатия сетчатки обоих глаз), невролога (спастический тетрапарез, грубая задержка психо-речевого, стато-моторного развития); выполнены УЗИ органов брюшной полости, почек (патологии не выявлено), ЭЭГ (локальные и пароксизмальные формы активности, а также эпилептиформная активность не выявляются). В отделении получал патогенетическую терапию, основу которой составила низкобелковая диета с низким содержанием триптофана и лизина (смесь аминокислот Нутриген из расчета 0,8г/кг/сутки), а также, согласно международным стандартам лечения данной патологии, высокодозное метаболическое лечение левокарнитином (150мг/кг/сутки), рибофлавином(100мг/сутки).
В возрасте 3 лет 4 месяцев (сентябрь 2019 г.) мальчик повторно госпитализирован в педиатрическое отделение № 2 НИИАП ФГБОУ ВО РостГМУ МЗ РФ для проведения очередного курса реабилитационной терапии с использованием вышеуказанных препаратов, а также применения ботулинотерапии с целью уменьшения проявлений спастико-гиперкинетического синдрома. После выписки продолжал получать метаболическую и диетотерапию.
Несмотря на постоянный пероральный прием вышеуказанных препаратов, при последней госпитализация в педиатрическое отделение № 2 в возрасте 3лет 10 месяцев ( в марте 2020 г.) массо-ростовые показатели (вес — 11,5 кг; рост — 95,0 см) не соответствовали средним величинам центильных таблиц, сохранялся спастическо-гиперкинетический синдром. Ребенок поступил в отделение в тяжелом состоянии, обусловленном метаболическим кризом, на фоне которого отмечались вялость, адинамия, бледность кожи, отсутствие аппетита и биохимические изменения: показатели гликемии — 0,6ммоль/л; лактата крови — 6,9ммоль/л, сочетавшиеся с гипокалиемией, ацидозом. После проведения инфузионной терапии растворами глюкозы с компонентами, введения левокарнитина, рибофлавина состояние стабилизировалось, показатели гликемии, лактата, электролитов и КОС нормализовались.
Таким образом, глутаровая ацидурия I типа относится к орфанным заболеваниям, касающимся наследственного дефицита глутарил-КоА-дегидрогеназы (GCDH). На примере клинического случая продемонстрирована сложность диагностического поиска, неблагоприятные особенности течения заболевания, видимо, обусловленные типом заболевания (мутацией с.1204С > Т (р.Аrg402Trp) в гетерозиготном состоянии) и поздно начатой терапией данному ребенку. Мы полагаем, что врачам педиатрического профиля следует иметь настороженность в отношении орфанных заболеваний, относящихся к группе ферментопатий. В этой связи следует помнить, что доказательным методом прижизненной диагностики глутаровой ацидурии I типа является тандемная масс-спектрометрия, которая позволяет выявить снижение концентрации свободного карнитина и резкое повышение концентрации глутарилкарнитина. Необходимо учитывать, что наиболее надежными, достоверными методами ранней диагностики глутаровой ацидурии I типа являются пренатальная или ранняя неонатальная диагностика (неонатальный скрининг). При их отсутствии следует учитывать данные нейросонографии, которые могут выявлять признаки макроцефалии и расширение ликворных пространств. Кроме того, важным для диагностики данного заболевания является оценка степени выраженности метаболического ацидоза у ребенка раннего возраста. Пациенты с субдуральными гематомами и / или битемпоральными арахноидальными кистами в сочетании с макроцефалией, дистониями, экстрапирамидными нарушениями с дебютом в раннем детском возрасте и характерными нейрорадиологическими изменения должны быть обследованы для исключения ГА-1.
В заключение следует обратить внимание, что фенотипический спектр нелеченной GA-1 варьируется от более распространенной формы (болезнь с младенческим началом) до менее распространенной формы (болезнь с более поздним началом, то есть после 6 лет и позже). Следует отметить, что фенотип GA-1 может широко варьироваться между необследованными членами семьи с одним и тем же генотипом, главным образом, в зависимости от возраста, в котором произошел первый острый энцефалопатический кризис: от трех месяцев до шести лет в GA-1с младенческим началом или после шести лет, особенно в позднем возрасте. Характерно, что кризисы приводят к острому двустороннему поражению головного мозга, прежде всего подкорковых структур и последующим сложным нарушениям движения. При проведении скрининга быстрое начало лечения бессимптомных форм означает, что большинство людей, у которых развились бы проявления GA-1 с детским или поздним началом, останутся без четкой симптоматики, поскольку необследованные случаи GA-1 с поздним началом могут проявляться как неспецифические неврологические нарушения (головные боли, головокружение, деменция и атаксия) [5], а также подвергаться повышенному риску других проявлений (например, почечной недостаточности), которые становятся очевидными по мере того, как понимание естественной истории лечения GA-1 продолжает развиваться [17][18].
Разработка и оценка планов лечения, помощи в обучении и воспитании пострадавших и их семьям, а также избежание побочных эффектов диетического лечения (то есть недоедания, нарушения роста) требуют многодисциплинарного подхода опытных специалистов из специализированного метаболического центра. Необходима регулярная оценка параметров роста и окружности головы, прогресса в развитии и образовательных потребностей, клинических признаков и симптомов двигательных расстройств, биохимических показателей и функции почек (у подростков и взрослых).
Следует помнить об оценке родственников группы риска: обследование всех сибсов группы риска любого возраста для ранней диагностики и лечения. Для новорожденных сибсов из группы риска в связи с тем, что в России пренатальное тестирование и программа глобального натального скрининга не проводятся, необходимо экстренное определение на семейные патогенные варианты GCDH (если они известны), либо срочное измерение мочевых органических кислот, аминокислот в плазме и профиль ацилкарнитина. Раннее назначение медикаментозной терапии в сочетании с низкобелковой и высокоуглеводной диетой позволяет уменьшить (а в отдельных случаях — предотвратить) необратимые повреждения структур головного мозга, в результате чего улучшить качество жизни данного контингента больных.
Финансирование. Исследование не имело спонсорской поддержки.
Finansing. The study did not have sponsorship.
Конфликт интересов. Авторы заявляют об отсутствии конфликта интересов.
Conflict of interest. Authors declares no conflict of interest
1. Herskovitz M., Goldsher D., Sela B.A., Mandel H. Subependymal mass lesions and peripheral polyneuropathy in adult‐ onset glutaric aciduria type. // I Neurology. – 2013. – V. 81. – P. 849-850. doi:10.1212/WNL.0b013e3182a2cbf2.
2. Kölker S., Valayannopoulos V., Burlina A.B., Sykut-Cegielska J., Wijburg F.A. et al. The phenotypic spectrum of organic acidurias and urea cycle disorders. Part 2: the evolving clinical phenotype. // J Inherit Metab Dis. – 2015. – V. 38. – P.1059– 1074. https://doi.org/10.1007/s10545-015-9840-x.
3. Михайлова С.В., Захарова Е.Ю., Петрухин А.С. Нейрометаболические заболевания у детей и подростков. Диагностика и подходы к лечению. М.: Литерра;2019.
4. Токарева Ю.В., Котов А.С., Сидоров О.П. Генетически подтвержденная L-2-гидроксиглутаровая ацидурия. // Неврологический журнал. – 2017. –Т.22, № 1. – С.45-49. doi: 10.18821/1560-9545-2017-22-1-45-49.
5. Boy N., Mengler K., Thimm E., Schiergens K.A., Marquardt T. et al. Newborn screening: a disease-changing intervention for glutaric aciduria type 1. // Ann Neurol. – 2018. – V. 83. – P. 970–979. doi: 10.1002/ana.25233.
6. Boy N., Mühlhausen C., Maier E.M., Heringer J., Assmann B. et al. Proposed recommendations for diagnosing and managing individuals with glutaric aciduria type I: second revision. // J Inherit Metab Dis. – 2017. – V. 40. – P. 75-101. doi: 10.1007/s10545-016-9999-9.
7. Tsai F.C., Lee H.J., Wang A.G., Hsieh S.C., Lu Y.H. et al. Experiences during newborn screening for glutaric aciduria type 1: diagnosis, treatment, genotype, phenotype, and outcomes. // J Chin Med Assoc. – 2017. – V. 80. P. 253-261. doi: 10.1016/j.jcma.2016.07.006.
8. Михайлова С.В., Захарова Е.Ю., Бобылова М.Ю., Ильина Е.С., Банин А.В. и др. Глутаровая ацидурия тип 1: клиника, диагностика и лечение // Журнал неврологии и психиатрии им. С.С. Корсакова. – 2007. – № 10. – С. 4-12. eLIBRARY ID: 9597007
9. Kölker S., Garcia-Cazorla A., Valayannopoulos V., Lund A.M., Burlina A.B. et al. The phenotypic spectrum of organic acidurias and urea cycle disorders. Part 1: the initial presentation. // J Inherit Metab Dis. – 2015. – V. 38. – P. 1041-1057. doi: 10.1007/s10545-015-9839-3
10. Boy N., Heringer J., Brackmann R., Bodamer O., Seitz A. et al. Extrastriatal changes in patients with late-onset glutaric aciduria type I highlight the risk of long-term neurotoxicity. // Orphanet J Rare Dis. – 2017. – V. 12. – P. 77. doi: 10.1186/s13023-017-0612-6.
11. Harting I., Boy N., Heringer J., Seitz A., Bendszus M. et al (1)H‐MRS in glutaric aciduria type 1: Impact of biochemical phenotype and age on the cerebral accumulation of neurotoxic metabolites. // J Inherit Metab Dis. – 2015. – V. 38. – P. 829–838. https://doi.org/10.1007/s10545-015-9826-8.
12. Баранов А.А., Намазова-Баранова Л.С., Боровик Т.Э., Бушуева Т.В., Глоба О.В. и др. Глутаровая ацидурия тип I у детей. Клинические рекомендации союза педиатров России. – М., 2017.
13. Kranendijk M., Struys E.A., Salomons G.S., Van der Knaap M.S., Jakobs C. Progress in understanding 2-hydroxyglutaric acidurias. // J. Inherit. Metab. Dis. – 2012. – V. 35, № 4. – P. 571-587. doi: 10.1007/s10545-012-9462-5.
14. Steenweg M.E., Jakobs C., Errami A., van Dooren S.J., Adeva Bartolomé M.T., et al. An overview of L-2-hydroxyglutarate dehydrogenase gene (L2HGDH) variants: a genotypephenotype study. // Hum. Mutat. – 2010. – V. 31, №4. – P. 380-390. doi: 10.1002 /humu.21197.
15. Mohammad S.A., Abdelkhalek H.S., Ahmed K.A., Zaki OK. Glutaric aciduria type 1: neuroimaging features with clinical correlation. // Pediatr Radiol. – 2015. – V. 45. – P. 1696-1705. https://doi.org/10.1007/s00247-015-3395-8.
16. Boy N., Mühlhausen C., Maier E.M., Heringer J., Assmann B. et al. Additional individual contributors. Proposed recommendations for diagnosing and managing individuals with glutaric aciduria type I: second revision. // J Inherit Metab Dis. – 2017. – V. 40, № 1. – P. 75-101. doi: 10.1007/s10545-016-9999-9.
17. Larson A., Goodman S. Glutaric Acidemia Type 1. 2019 Sep 19. // Adam M.P., Ardinger H.H., Pagon R.A. et al., editors. GeneReviews®. – University of Washington, Seattle; 1993-2020. PMID: 31536184.
18. Peters V., Morath M., Mack M., Liesert M., Buckel W. et al. Formation of 3-hydroxyglutaric acid in glutaric aciduria type I: in vitro participation of medium chain acyl-CoA dehydrogenase. // JIMD Rep. – 2019. – V. 47, № 1. – P. 30-34. doi: 10.1002/jmd2.12026.
д.м.н., проф., проректор по акушерству и педиатрии (директор НИИАП),
Ростов-на-Дону
д.м.н., проф., главный научный сотрудник педиатрического отдела,
Ростов-на-Дону
к.м.н., педиатрическое отделение № 2, врач-педиатр,
Ростов-на-Дону
к.м.н., педиатрическое отделение № 2, заведующая,
Ростов-на-Дону
к.м.н., амбулаторноконсультативное отделение, врач-невролог,
Ростов-на-Дону
к.м.н., педиатрическое отделение № 1, заведующая,
Ростов-на-Дону
педиатрическое отделение № 2, врач-педиатр,
Ростов-на-Дону
отделение лучевой диагностики, заведующая,
Ростов-на-Дону
к.м.н., педиатрическое отделение № 2, врач-педиатр,
Ростов-на-Дону
Лебеденко А.А., Бережанская С.Б., Тодорова А.С., Вострых Н.Н., Каушанская Е.Я., Лукьянова Е.А., Папшева Е.А., Смыкова Г.Н., Тараненко Л.Н. Редкий случай глутаровой ацидурии I типа у ребенка раннего возраста. Медицинский вестник Юга России. 2020;11(4):84-91. https://doi.org/10.21886/2219-8075-2020-11-4-84-91
Lebedenko A.A., Berezhanskay S.B., Todorova A.S., Vostrykh N.N., Kaushanskay E.Y., Lukyanova E.A., Papsheva E.A., Smykova G.N., Taranenko L.N. A rare case of type i glutaric aciduria in an early child. Medical Herald of the South of Russia. 2020;11(4):84-91. (In Russ.) https://doi.org/10.21886/2219-8075-2020-11-4-84-91
344022, г. Ростов-на-Дону, пер. Нахичеванский, 29
Ростовский государственный медицинский университет
Тел.: +7 918 571 0558
E-mail: journal@medicalherald.ru


































