



Contents
Scroll to:
https://doi.org/10.21886/2219-8075-2023-14-1-38-42
Scroll to:
Disorder of sex development (DSD) is a term used to refer to congenital disorders that led to atypical structure of the genitals. The cause of DSD is a disorder of the embryonic development of the reproductive system due to chromosomal, genetic pathology or other adverse effects on pregnancy. DSD entails difficulties with social adaptation of the family, leads to severe psychological disorders in the child and his relatives. Sex of a child with DSD should be established only after a full examination and consultation of specialists in this field. A clinical case is presented to illustrate the complexity of differential diagnosis and choice of passport sex in a child with one of the rare forms of DSD.
Shaydullina M.R., Akramov N.R., Valeeva F.V., Alimetova Z.R., Kolbasina E.V. Clinical сase of disorder of sex development with karyotype 47XYY. Medical Herald of the South of Russia. 2023;14(1):38-42. (In Russ.) https://doi.org/10.21886/2219-8075-2023-14-1-38-42
The term “Disorders of sexual development” (DSD) is currently considered the most appropriate term to describe diseases characterized by a mismatch between the structure of external and internal genitalia and genetic sex [1]. When such a condition is detected in a child in clinical practice, further tactics of doctors depend on a complete, reliable, and timely examination. The choice of sex in this case depends on many criteria (results of genetic, hormonal, ultrasound, and other studies), as well as on the patient’s ability for further reproductive function, the possibility of adequate medical intervention and further socialization of the individual. DSD of the child is an important psychological problem for parents, who should also be involved in the choice of sex.
Violations of sexual differentiation are conditionally divided into three large groups: DSD 46,XY, DSD 46,XX, and sex chromosomal disorders, in which various variants of abnormal karyotype are registered.
Jacobs syndrome (47XYY males) was first described in 1960 [2] and is currently not considered a rare pathology: according to various sources, the 47XYY karyotype is reported with a frequency of 1 in 1,000 [3] to 1 in 10,000 men [4]. However, up to 85% of patients remain undetected [2].
This aneuploidy is associated with phenotypic features such as high stature, macrocephaly, macroorchidism, hypertelorism, and clinodactyly [2]. Patients with karyotype 47XYY were described to have delayed motor development with muscle hypotonia and limb tremor [2][3]. However, most often, patients with Jacobs syndrome attract attention with impaired neurocognitive functions (dyslexia, dysgraphia, hyperactivity syndrome, various autism spectrum disorders, and impaired behavioral reactions leading to the social disadaptation of patients) [2][3][5]. Despite the presence of isolated descriptions of anomalies of urogenital tract development in the form of microphallus, cryptorchidism, hypospadias, and delayed sexual development in boys with karyotype 47XYY [2], studies of adult patient populations indicate normal or high testosterone levels [6]. This abnormality is most often diagnosed in men during infertility examinations [7] or “whole-body” karyotyping in prisons and psychiatric clinics [2][5].
This clinical case is a rare example of a sex development disorder associated with karyotype 47 XYY and reflects the difficulties in choosing the passport sex in children with genital abnormalities.
Child S. was born from the third pregnancy, which was accompanied by acute respiratory infections in the first trimester, threatened termination of pregnancy at 29 weeks, and second term labor. Growth at birth was 54 cm, weight was 3,450 g, 7–8 points on the Apgar scale. Due to the irregular structure of external genitalia, he was transferred to the neonatal pathology department, and then, to the endocrinology department for further examination with the diagnosis “Disorder of sexual development. Neonatal jaundice. Intrauterine infection of unspecified etiology. Urinary tract infection. Perinatal damage to the central nervous system. Small anomalies of heart development (open oval window)”.
On the first day of life, the level of 17-OH-progesterone (6.0 nmol/L, with the norm (N) up to 15 nmol/L) was examined to exclude a life-threatening condition of a salt-losing form of adrenogenital syndrome. Ultrasound investigation showed that at the age of 1 week, the right testicle was visualized in the projection of the middle third of the inguinal canal, the left testicle was not reliably determined, the uterus was found behind the bladder. Karyotyping at the age of 1 week revealed chromosomal abnormality – 47, XYY. Repeated determination of karyotype at the age of 2 weeks revealed mosaicism on sex chromosomes 47, XYY (142)/45, X0 (8).
Endocrine status evaluation performed at the age of 1 month confirmed the presence of testicular tissue in the child: basal levels of testosterone, antimullerian hormone (AMH), inhibin B (Table 1), positive test with chorionic gonadotropin (testosterone initially – 6.94 nmol/l, 24 hours after injection #3 of 250 IU – 13.87 nmol/l). The parents’ decision to choose male passport sex was therefore supported.
Table 1
The results of the examination of the child at the age of 1 and 16 months
|
Unit parameter |
Level at 1 month of age |
Level at 16 months of age |
Reference interval |
|
FSH (mIU/ml) |
7.54 |
5.8 |
men 0–1 year <3.5 women 0–1 year 1.84–20.26 |
|
LH (mIU/ml) |
3.6 |
1.1 |
0–1 year <6.34 |
|
Testosterone (nmol/l) |
6.1 |
˂0.025 |
men 4 days–6 months 0.30–10.36 women 4 days–9 years <2.15 |
|
Estradiol (pg/ml) |
˂5 |
men 0–1 year <86 women 0–1 year <155 |
|
|
AMH (ng/ml) |
16.91 |
0.8 |
pre–pubertal men 3.80–159.80 pre–pubertal women <8.90 |
|
Inhibin B (pg/ml) |
65.95 |
3.2 |
men 0–18 years 4.0–352.0 women 0–18 years <83.0 |
At the age of 1 year and 4 months, the child was again admitted to the endocrinology department of a children's multidisciplinary hospital for preparation for genital masculinizing plastic surgery.
The objective examination showed that the boy had an abnormal structure of the external genitalia: hypospadias, scrotal form; split scrotum; bilateral cryptorchidism, microphallus (2.0 cm). The external urethral orifice opened at the base of the penis between the parts of the split scrotum (Fig. 1). The gonads were not palpated in the scrotum.
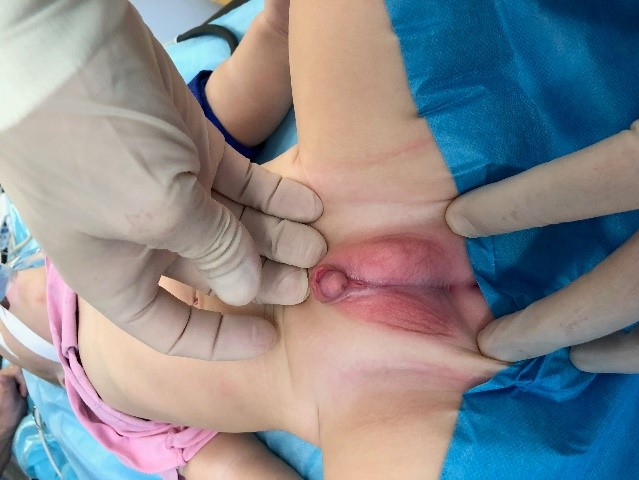
Figure 1a. The appearance of the external genitalia

Fig. 1b. Scrotal hypospadias
The study of hormone levels showed a significant dynamic decrease not only in testosterone levels, which is natural at the age of minipuberty, but also in AMH and Inhibin B.
The child underwent a repeated study of karyotype by the FISH method, the absence of mosaicism was established, and the presence of disomy on the Y chromosome was confirmed (47, XYY – 100%). Due to the assumption of complete absence or disturbances in the structure of the SRY (Sex-determining Region Y) gene, the sequence of Y-chromosome SRY was evaluated, pathogenic and probably pathogenic variants of SRY gene mutations were not found. Molecular genetic diagnosis (panel “Disorder of sexual development” of 38 genes) was carried out; no pathogenic and probably pathogenic variants, as well as variants with unknown clinical significance, explaining the cause of the disease at the molecular genetic level, were found.
In order to determine the plan of surgical intervention, panendoscopy (cystourethroscopy, colposcopy) was performed: a vaginal entrance was found between the external and internal sphincters of the urogenital tract; the vagina was 6 cm long (Fig. 2).
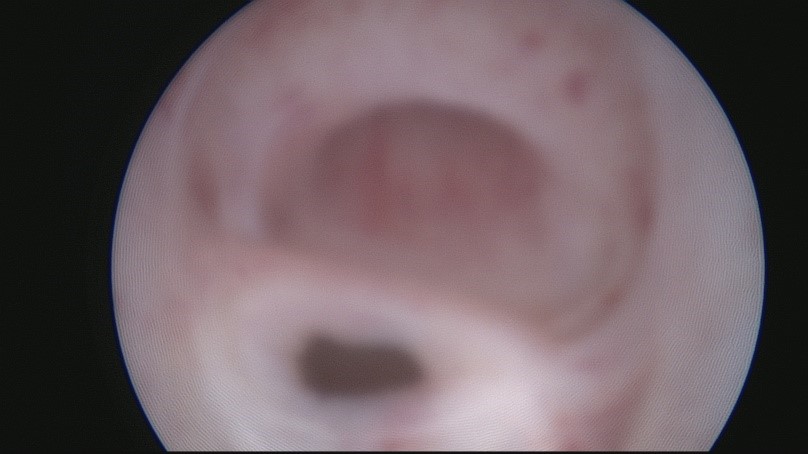
Fig. 2. Cystoscopy
Laparoscopic revision of the abdominal cavity revealed the following: the left gonad was round, the appendage was hyperemic, dense. The right gonad was at the end of the fallopian tube, dysplasic. The appearance of both gonads was not typical of either testis or ovary (Fig. 3a, 3b). The left inguinal ring was not obliterated. There was a uterus of small size.

Fig. 3a. Appearance of dysplastic gonads: the left gonad with a diameter of 1 cm
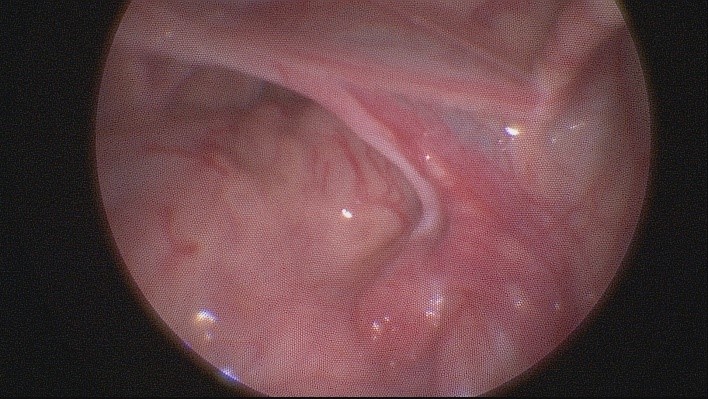
Fig. 3b. Appearance of dysplastic gonads: the right gonad
The council of specialists who participated in the examination of the child made a diagnosis of "Sexual development disorder; sex chromosomal anomalies 47, XYY; gonadal dysgenesis". Due to the absolute indications for gonadectomy (dysgenesis, the location of the gonads in the abdominal cavity is associated with a high risk of malignization [8]), the presence of a uterus, vagina, and the possibility of one-stage feminizing genital plastic surgery, the patient was recommended to change his passport sex to female. If the family decides to choose male passport sex, gonadectomy and multistage masculinizing external genital plasty are recommended.
Despite warnings about possible risks and complications from the upcoming medical interventions, recommendations of a psychologist and other specialists, the parents decided to keep the child’s male sex.
Analysis of available publications (sufficient masculinization of external genitalia at birth in the vast majority of boys with karyotype 47, XYY), the absence of identified Y-chromosome anomalies and mutations explaining the cause of the disease in this patient, did not allow the authors to exclude the possibility of teratogenic effects that led to the formation of dysgenesis of the gonads in early pregnancy, insufficient intrauterine secretion of AMH (presence of uterus, fallopian tubes) and testosterone, which led to the incorrect development of the child’s sex.
1. Hughes IA, Houk C, Ahmed SF, Lee PA; Lawson Wilkins Pediatric Endocrine Society/European Society for Paediatric Endocrinology Consensus Group. Consensus statement on management of intersex disorders. J Pediatr Urol. 2006;2(3):148-62. DOI: 10.1016/j.jpurol.2006.03.004
2. Bloy L, Ku M, Edgar JC, Miller JS, Blaskey L, Ross J, Roberts TPL. Auditory evoked response delays in children with 47,XYY syndrome. Neuroreport. 2019;30(7):504-509. DOI: 10.1097/WNR.0000000000001233
3. Stochholm K, Juul S, Gravholt CH. Socio-economic factors affect mortality in 47,XYY syndrome-A comparison with the background population and Klinefelter syndrome. Am J Med Genet A. 2012;158A(10):2421-9. DOI: 10.1002/ajmg.a.35539
4. Berglund A, Viuff MH, Skakkebæk A, Chang S, Stochholm K, Gravholt CH. Changes in the cohort composition of turner syndrome and severe non-diagnosis of Klinefelter, 47,XXX and 47,XYY syndrome: a nationwide cohort study. Orphanet J Rare Dis. 2019;14(1):16. DOI: 10.1186/s13023-018-0976-2
5. Green T, Flash S, Reiss AL. Sex differences in psychiatric disorders: what we can learn from sex chromosome aneuploidies. Neuropsychopharmacology. 2019;44(1):9-21. DOI: 10.1038/s41386-018-0153-2
6. Ross JL, Roeltgen DP, Kushner H, Zinn AR, Reiss A, et al. Behavioral and social phenotypes in boys with 47,XYY syndrome or 47,XXY Klinefelter syndrome. Pediatrics. 2012;129(4):769-78. DOI: 10.1542/peds.2011-0719
7. Borjian Boroujeni P, Sabbaghian M, Vosough Dizaji A, Zarei Moradi S, Almadani N, et al. Clinical aspects of infertile 47,XYY patients: a retrospective study. Hum Fertil (Camb). 2019;22(2):88-93. DOI: 10.1080/14647273.2017.1353143
8. Fallat ME, Donahoe PK. Intersex genetic anomalies with malignant potential. Curr Opin Pediatr. 2006;18(3):305-11. DOI: 10.1097/01.mop.0000193316.60580.d7
Maria R. Shaydullina, Cand. Sci. (Med.), associate professor of the Department of Endocrinology; head doctor pediatric endocrinologist
Kazan
Authors declares no conflict of interest.
Nail R. Acramov, Dr. Sci. (Med.), Professor of the Department of Pediatric Surgery
Kazan
Authors declares no conflict of interest.
Farida V. Valeeva, Dr. Sci. (Med.), Professor, head of Department of Endocrinology
Kazan
Authors declares no conflict of interest.
Zulfiia R. Alimetova, MD, assistant of the Department of Endocrinology
Kazan
Authors declares no conflict of interest.
Elena V. Kolbasina, Head of the Department of Endocrinology
Nizhny Novgorod
Authors declares no conflict of interest.
Shaydullina M.R., Akramov N.R., Valeeva F.V., Alimetova Z.R., Kolbasina E.V. Clinical сase of disorder of sex development with karyotype 47XYY. Medical Herald of the South of Russia. 2023;14(1):38-42. (In Russ.) https://doi.org/10.21886/2219-8075-2023-14-1-38-42
29, Nakhichevansky Lane, Rostov-on-Don, 344002
Rostov State Medical University
Тel.: +79185710558
e-mail: journal@medicalherald.ru


































