



Содержание
Перейти к:
https://doi.org/10.21886/2219-8075-2023-14-4-35-43
Перейти к:
Дефицит GATA2 – редкое заболевание, относящееся к группе врождённых дефектов фагоцитов, которое клинически проявляется четырьмя синдромами: синдромом MonoMAC (миедисплазия и иммунодефицит, ассоциированный с развитием инфекций, вызванных Mycobacterium avium complex), синдромом дефицита моноцитов, дендритных клеток, В- и NK-лимфоцитов, синдромом Эмбергера, включающим первичную лимфедему с миелодисплазией и нейросенсорную потерю слуха, а также семейным миелодиспластическим синдромом и острым миелоидным лейкозом. Заболевание наследуется по аутосомно-доминантному типу, но в большинстве случаев мутации зародышевой линии гена GATA2 возникают de novo. Первые проявления заболевания встречаются в раннем взрослом возрасте, течение дефицита GATA2 является вариабельным и может различаться у лиц в одной семье, имеющих схожие генетические варианты. В статье представлен клинический случай манифестации дефицита GATA2 в возрасте семи лет в виде развития генерализованного веррукоза, лимфостаза нижней конечности, генерализованного туберкулёза с поражением брюшной полости, малого таза, органов грудной клетки. В результате обследования выявлен дефицит моноцитов, В- и NK-лимфоцитов, миелодиспластический синдром с мультилинейной дисплазией. Представлено подробное описание клинической картины и особенностей течения первичного иммунодефицитного состояния, результаты проведенного обследования и лечения.
Фролов Е.А., Абдулаева Ф.И., Горностаева Ю.А., Латышева Т.В., Латышева Е.А., Аминова Г.Э. Особенности клинической картины и течения дефицита GATA2, осложнённого генерализованным веррукозом с исходом в миелодиспластический синдром во взрослом возрасте. Медицинский вестник Юга России. 2023;14(4):35-43. https://doi.org/10.21886/2219-8075-2023-14-4-35-43
Frolov E.A., Abdulaeva F.I., Gornostaeva U.A., Latysheva T.V., Latysheva E.A., Aminova G.E. Features of the clinical picture and course of GATA2 deficiency complicated by generalized verrucosis with an outcome in myelodysplastic syndrome in adulthood. Medical Herald of the South of Russia. 2023;14(4):35-43. (In Russ.) https://doi.org/10.21886/2219-8075-2023-14-4-35-43
В мировой литературе представлено более 480 различных форм врождённых ошибок иммунной системы (первичных иммунодефицитов — ПИД), которые в зависимости от поражённого звена иммунной системы разделены на 10 групп [1]. Клиническая картина ПИД вариабельна, характеризуясь повышенной восприимчивостью к инфекциям, аутоиммунным и аутовоспалительным синдромами, патологической лимфопролиферацией, недостаточностью костного мозга и/или онкологическим заболеваниям [2][3]. Одним из представителей первичных иммунодефицитных состояний, сочетающим в себе многообразие клинических проявлений, является дефицит GATA2, который, согласно международной классификации, включён в группу врождённых дефектов количества или функций фагоцитов.
История открытия дефицита GATA2 относится к 2010 г., когда группой учёных из США [4] впервые был описан синдром иммунодефицита и миелодисплазии, проявляющийся глубоким дефицитом моноцитов в периферической крови и склонностью к развитию инфекций, вызванных Mycobacterium avium complex, который был сокращённо назван MonoMAC. В то же время было описано три схожих синдрома: синдром дефицита моноцитов, дендритных клеток, В- и NK-лимфоцитов, Синдром Эмбергера, включающий первичную лимфедему с миелодисплазией и нейросенсорную потерю слуха, а также семейный миелодиспластический синдром (МДС), острый миелоидный лейкоз [5–7]. В 2011 г. было выяснено, что все четыре синдрома являются результатом мутаций зародышевой линии гена GATA2 и представляют собой разнообразные проявления одного и того же заболевания, впоследствии названного дефицитом GATA2.
Ген GATA2 расположен на длинном плече третьей хромосомы, механизм наследования заболевания аутосомно-доминантный [8]. Белок GATA2 принадлежит к семейству ядерных факторов транскрипции, которые связываются с консенсусной последовательностью ДНК и другими факторами транскрипции, регулируя экспрессию множества генов-мишеней [9]. У человека он экспрессируется в гематологических клетках на стадии стволовой клетки и более поздних стадиях развития клеток-предшественников. Повышение и/или снижение экспрессии гена регулируют прогрессирование этих незрелых клеток в их конечные формы, а также определённые типы тканевых клеток, такие как макрофаги и тучные клетки [10].
На сегодняшний день в мире зарегистрировано около 500 пациентов с дефицитом GATA2 [11] и описано более 150 различных патогенных вариантов гена, которые в большинстве случаев возникают de novo [12]. Течение дефицита GATA2 вариабельно и может различаться у лиц в одной семье, имеющих схожие генетические варианты. Наиболее часто первые проявления заболевания встречаются в раннем взрослом возрасте. Фенотип заболевания гетерогенен, не имеет чёткой корреляции с генотипом и характеризуется неполной клинической пенетрантностью [13]. Симптомы могут включать тяжёлые рецидивирующие (вирусные, бактериальные, грибковые) инфекции, цитопении, лимфедему, альвеолярный протеиноз и злокачественные миелоидные заболевания [14][15]. Инфекционные осложнения при дефиците GATA2 являются одной из основных причин инвалидизации данных пациентов. Они связаны с дефицитом B-лимфоцитов, NK-клеток, а также с дефектами врождённого иммунного ответа, включая нарушение продукции интерферона I типа [16] и характеризуются повышенной восприимчивостью к вирусным инфекциям (например, инфекциям, вызванным вирусом папилломы человека (ВПЧ) и простого герпеса, нетуберкулезным микобактериям и к другим более распространённым бактериальным респираторным инфекциям). Потеря слуха также встречается в симптомокомплексе дефицита GATA2 и обусловлена участием гена в вестибулярном морфогенезе полукружных каналов формировании перилимфатического пространства вокруг полукружных каналов внутреннего уха [17].
Единственным куративным методом лечения дефицита GATA2 на сегодняшний день является аллогенная трансплантация гемопоэтических стволовых клеток (ТГСК). Как и в большинстве случаев ПИД, решение вопроса о проведении ТГСК является серьёзной задачей для врачей онкологов-гематологов и требует комплексной оценки текущего состояния пациента. В иностранной литературе представлены данные о 86% выживаемости на протяжении двух лет после ТГСК (n = 22, средний возраст — 26 лет) [18], 57% выживаемости через 3,5 года после ТГСК (n = 14, средний возраст — 33 года) [19], 72 %, 65% и 54% выживаемости через 1, 2 и 4 года после ТГСК соответственно (n = 21, возраст пациентов — от 15 до 48 лет) [14], однако данные когорты не могут быть сопоставимы между собой в связи с различиями по тяжести клинических проявлений заболевания и режимам кондиционирования.
Пациентка Ш., 31 год. Поступила в отделение с жалобами на неустойчивый стул со склонностью к диарее, жжение и болезненность при мочеиспускании, рецидивирующие бородавки.
В детстве росла и развивалась согласно возрастным нормам. Семейный анамнез не отягощён, однако у дедушки по материнской линии отмечались жалобы на значительное снижение слуха. В период с 3 до 18 лет возникали частые нетяжёлые обострения отита после перенесённых ОРВИ (до четырёх раз в год), лечение — местное и симптоматическое с положительным эффектом.
С 7 лет на коже кистей впервые стали возникать простые бородавки, с 24 лет (2016 г.) — кондиломы в области гениталий. Количество бородавок с возрастом увеличивалось в размере и в количестве (рис. 1, 2). Проводились неоднократные курсы наружной терапии, системная иммуномодулирующая терапия без видимого улучшения. Удаление бородавок методом криодеструкции было неэффективно, однако в местах, где применялся радиоволновой метод удаления, новые элементы не возникали, но впоследствии оставались келоидные рубцы.

Рисунок 1. Пациентка Ш., 31 год, диагноз —
«Первичное иммунодефицитное состояние, дефицит GATA2.
Генерализованный веррукоз, преимущественно акральной локализации».
Figure 1. Patient Sh., 31 years old, diagnosis —
“Primary immunodeficiency, GATA2 deficiency.
Generalized verrucosis, predominantly acral localization”.
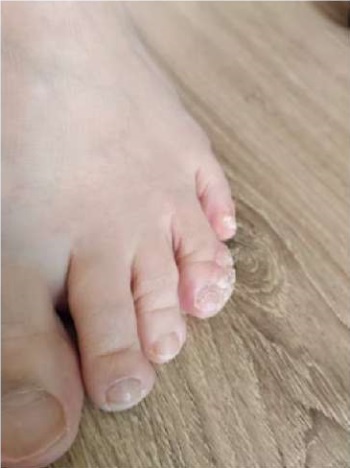
Рисунок 2. Пациентка Ш., 31 год, диагноз —
«Первичное иммунодефицитное состояние, дефицит GATA2.
Вульгарные плантарные бородавки».
Figure 2. Patient Sh., 31 years old, diagnosis —
“Primary immunodeficiency, GATA2 deficiency.
Vulgar plantar warts”.
С 11 лет развился и стал прогрессировать лимфостаз правой нижней конечности неуточнённого генеза, в 20 лет проведена операция с формированием лимфовенозного анастомоза без существенного эффекта (рис. 3).
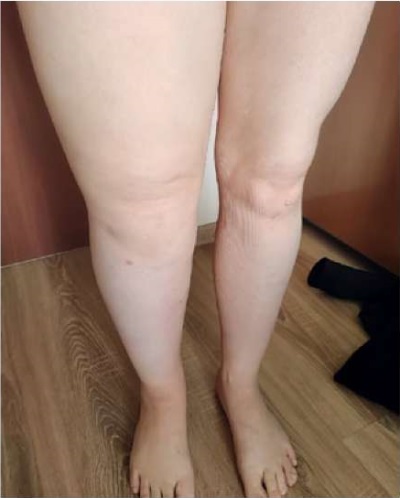
Рисунок 3. Пациентка Ш., 31 год, диагноз —
«Первичное иммунодефицитное состояние: дефицит GATA2.
Лимфостаз правой нижней конечности».
Figure 3. Patient Sh., 31 years old, diagnosis —
“Primary immunodeficiency, GATA2 deficiency.
Lymphostasis of the right lower limb”.
В 15 лет — генерализованный туберкулез с поражением брюшной полости, малого таза, органов грудной клетки. Mycobacterium tuberculosis выявлена при бактериологическом исследовании плевральной жидкости, в отделяемом из абсцесса в малом тазу, в отделяемом из тонкокишечных свищей. В течение 2 лет проводилась стационарная консервативная терапия, повторные хирургические вмешательства (резекция поперечной ободочной кишки, резекция восходящей ободочной кишки, тубэктомия справа по поводу туберкулеза малого таза (лапаротомия), вскрытие и дренирование абсцесса малого таза — рис. 4). В период с 15 до 30 лет состояла на учёте в противотуберкулезном диспансере, сохранялся отрицательный результат диаскинтеста. С декабря 2022 г. снята с учёта, констатирована клинико-лабораторная ремиссия туберкулеза.
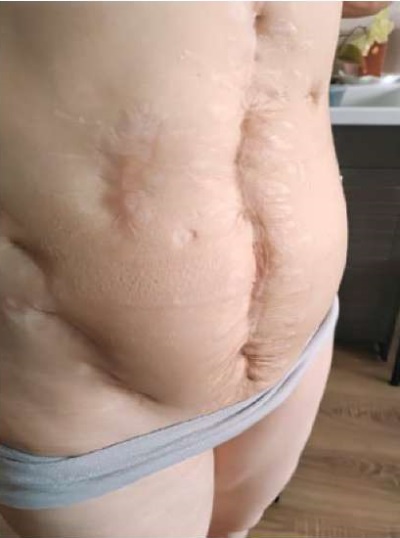
Рисунок 4. Пациентка Ш., 31 год, диагноз —
«Первичное иммунодефицитное состояние, дефицит GATA2.
Туберкулёзное поражение лёгких, висцеральный туберкулёз,
генитальный туберкулёз». Состояние после хирургического лечения (2007–2009 гг.).
Figure 4. Patient Sh., 31 years old, diagnosis — “Primary immunodeficiency,
GATA2 deficiency. Tuberculous lung lesion, visceral tuberculosis,
genital tuberculosis”. Condition after surgical treatment (2007–2009).
С 16 лет (2009 г.) в анализах выявлена анемия смешанного генеза (железодефицитная и пернициозная). В клиническом анализе крови впервые выявлена лейкопения 3,8×10^9/л.
В 18 лет в КВКД№1 проведено исследование иммунного статуса: иммуноглобулина А (IgA) — 1.7 г/л (норма — 0.9–5.0); IgG 30 г/л (норма — 9–20); IgМ — 4.9 г/л (норма — 0.7–3.7); лейкоциты — 3,4×10^9/л, лимфоциты — 0.9×10^9/л, CD3+ Т-лимфоциты — 611 кл/мкл (норма — 800–2200), CD4+ Т-лимфоциты — 275 кл/мкл (норма — 600–1600), CD8+ Т-лимфоциты — 321 кл/мкл (норма — 300–800), CD19+ В-лимфоциты — 19 кл/мкл (норма — 100–500).
В 22 года пациентка была определена на стационарное лечение в МНПЦ борьбы с туберкулёзом в связи с жалобами на боль в пояснице. Данных о туберкулёзе мочевой системы не выявлено, диаскинтест — отрицательный. При бактериологическом исследовании мочи выявлен рост Proteus mirabilis 10^5. Согласно результатам клинического анализа крови, гемоглобин — 116 г/л, лейкоциты — 3.2×109/л, лимфоциты — 0.8×109/л, гранулоциты — 2.2×109/л, моноциты — 0,064×109/л, тромбоциты — 243×109/л. По данным ультразвукового исследования органов брюшной полости, обнаружены умеренная гепатоспленомегалия, кальцинаты печени, селезёнки.
В 29 лет пациентка перенесла вирусную инфекцию COVID-19 тяжёлого течения, подтверждённую ПЦР-исследованием отделяемого из зева/носоглотки, и со степенью поражения лёгких КТ-2. В анализах отмечалось повышение с-реактивного белка до 186 мг/дл. Согласно результатам клинического анализа крови, гемоглобин — 123 г/л, эритроциты — 4.53×10^12/л, лейкоциты — 3.73×10^9/л, нейтрофилы — 3.07×10^9/л, лимфоциты — 0.24×10^9/л, тромбоциты — 179×10^9/л.
В 30 лет (2023 г.) пациентка отправлена на стационарное лечение в отделение онкогинекологии УКБ №4. Удалены полип эндометрия, левая наружная половая губа, кондиломы генитальной области. При гистологическом исследовании выявлена карцинома вульвы in situ (Т0-1N0M0). Проведено МРТ органов малого таза: обнаружены увеличенные лимфоузлы и очаговые изменения костей таза, макропризнаки опухолевого поражения вульвы и влагалища не выявлено.
В 31 год пациентка впервые обратилась к иммунологу. При обследовании в ФГБУ ГНЦ Институт иммунологии ФМБА России, по результатам молекулярно-генетического исследования (полное секвенирование экзома), обнаружен ранее не описанный, вероятно, патогенный вариант с.386del в гетерозиготном состоянии в экзоне 3 из 6 гена GATA2, в гетерозиготном (доминантном) состоянии, приводящий к сдвигу рамки считывания p.Ser129Ter.
Рост — 165 см, масса тела — 67 кг. Состояние — стабильное, тяжёлое — по основному заболеванию. Сознание — ясное. Кожные покровы: множественные веррукозные элементы на коже тыльной и ладонной поверхности кистей, пальцев рук, вульгарные бородавки на коже стоп (рис. 4, 5), единичные веррукозные элементы на коже лица (лобная, носовая область), нижних конечностей. Множественные келоидные рубцы в урогенитальной области. Рубцы в области живота, поясницы, после перенесённых лапароскопических операций (рис. 7). Лимфостаз правой нижней конечности (рис. 6). Носовое дыхание — свободное. Дыхание — жёсткое, хрипы не выслушиваются. Тоны сердца — ясные, ритм — правильный. Показатели гемодинамики — стабильные.
В клиническом анализе крови отмечалась анемия: гемоглобин — 85 г/л, эритроциты — 3.03×10^12/л, лейкопения, лимфопения (лейкоциты — 2.1×10^9/л, нейтрофилы — 1.7×10^9/л, лимфоциты — 0.31×10^9/л, моноциты — 0,01×10^9/л), тромбоциты — 179×10^9/л, незначительное повышение СОЭ по Вестергрену — до 30 мм/ч.
В биохимическом анализе крови обнаружено снижение уровня общего белка до 50 г/л (66–83 г/л), креатинин — 50 мкмоль/л (58–96 мкмоль/л), железо — 2,2 мкмоль/л (10,7–32,2 мкмоль/л), ферритин —26 нг/мл (11–306,8 нг/мл), С-реактивный белок — 6.6 мг/дл (<5 мг/дл).
В иммунологическом обследовании обращало на себя внимание снижение иммуноглобулина А (IgA) до 69 мг/дл (норма — 100–350), IgG — 528 мг/дл (норма — 900–1800) при нормальном IgМ — 89 мг/дл (норма — 80–250), резкое снижение CD3+ Т-клеток до 177 кл/мкл (норма — 800–2200), а также CD3+CD4+ Т-хелперы — 68 кл/мкл (норма — 600–1600), CD3+CD8+ Т-цитотоксические — 97 кл/мкл (норма — 190–650), CD3-CD16,56+ NK-клетки — 115 кл/мкл (норма — 150–600), CD19+ B-клетки — 0,4 кл/мкл (норма — 100–500) (табл. 1). С учётом низкого количества В-лимфоцитов в периферической крови фенотипирование В-лимфоцитов не проводилось. Исследование фагоцитарной активности не проводилось в связи с отсутствием технической возможности в период госпитализации пациентки, оно будет проведено в дальнейшем.
Таблица / Table 1
Результаты иммунофенотипирования лимфоцитов периферической крови пациентки
Results of immunophenotyping of peripheral blood lymphocytes of the patient
Показатели/Indicators | Результат/Result | Норма/Normal |
Лимфоциты (на 1 мкл крови) Lymphocytes (per 1 μl of blood) | 300 | 1200–3000 |
CD3+ Т-клетки (% лимфоцитов) CD3+ T-cells (% lymphocytes) | 59,0 | 55–80 |
CD3+ Т-клетки (на 1 мкл крови) CD3+ T-cells (per 1 μl of blood) | 177 | 800–2200 |
CD3+CD4+ Т-хелперы (% лимфоцитов) CD3+CD4+ T-helpers (% lymphocytes) | 22,6 | 31–49 |
CD3+CD4+ Т-хелперы (на 1 мкл крови) CD3+CD4+ T-helpers (per 1 μl of blood) | 68 | 600–1600 |
CD3+CD8+ цитотоксические Т-клетки (% лимфоцитов) CD3+CD8+ cytotoxic T-cells (% lymphocytes) | 32,3 | 12–30 |
CD3+CD8+ цитотоксические Т-клетки (на 1 мкл крови) CD3+CD8+ cytotoxic T-cells (per 1 μl of blood) | 97 | 190–650 |
CD3+CD4+/CD3+CD8+ | 0,70 | 1,5–3,0 |
CD3+CD4+CD8+ (% лимфоцитов) CD3+CD4+CD8+ (% lymphocytes) | 1,6 | < 2 |
CD3-CD16,56+ NK-клетки (% лимфоцитов) CD3-CD16,56+ NK-cells (% lymphocytes) | 38,5 | 6–20 |
CD3-CD16,56+ NK-клетки (на 1 мкл крови) CD3-CD16,56+ NK-cells (per 1 μl of blood) | 115 | 150–600 |
CD19+ B-клетки (% лимфоцитов) CD19+ B-cells (% lymphocytes) | 0,14 | 5–19 |
CD19+ B-клетки (на 1 мкл крови) CD19+ B-cells (per 1 μl of blood) | 0,4 | 100–500 |
Согласно ПЦР-исследованию крови, ДНК цитомегаловируса, вируса Эпштейн-Барр, вируса герпеса 6 типа не обнаружена. Согласно ПЦР-исследованию соскоба из веррукозных элементов, обнаружено 5.9 копий ДНК HPV 16 типа на 10^5 клеток.
Согласно ПЦР-исследованию соскоба со слизистой в генитальной области (в количестве копий ДНК ВПЧ на 10^5 клеток), обнаружено HPV35 (7.8), HPV16 (8.0), HPV51 (7.9).
Согласно УЗИ региональных лимфоузлов, данных о патологически изменённых лимфоузлах не получено.
По данным УЗИ брюшной полости, выявлены кальцинаты в 4-м, 6-ом и 7-м сегментах печени размером 1,0 см, 3.5 см и 0,9 см соответственно, умеренная спленомегалия (длина — 143 мм, толщина — 48 мм).
Заключение КТ органов грудной клетки: посттуберкулезные, фиброзные, поствоспалительные изменения в лёгких; остаточные проявления интерстициальной, вероятно, вирусной пневмонии с признаками интерстициального фиброза; множественные очаги в лёгких, новый очаг в S10 нижней доли правого лёгкого; уплотнение (узловое образование?) в кортикальных отделах S10 нижней доли правого лёгкого (рис. 5, 6, 7).
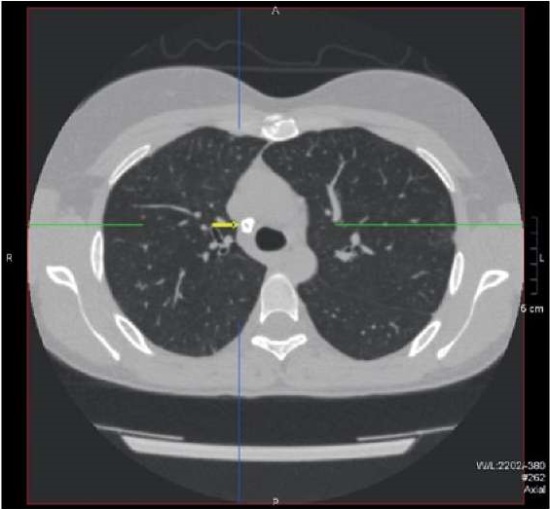
Рисунок 5. Пациентка Ш., 31 год, диагноз —
«Первичное иммунодефицитное состояние, дефицит GATA2.
Обызвествленный паратрахеальный лимфатический узел».
Figure 5. Patient Sh., 31 years old, diagnosis — “Primary immunodeficiency,
GATA2 deficiency. Calcified paratracheal lymph node”.
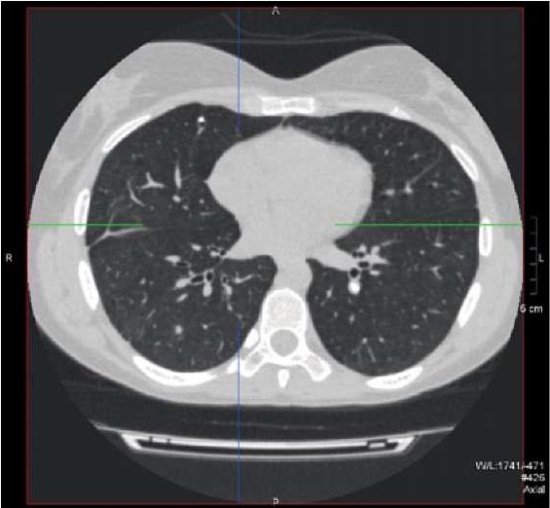
Рисунок 6. Пациентка Ш, 31 год, диагноз —
«Первичное иммунодефицитное состояние, дефицит GATA2.
Обызвествленный лимфатический узел в корне левого лёгкого.
Обызвествленный очаг и мягкотканый очаг в правом лёгком».
Figure 6. Patient Sh., 31 years old, diagnosis — “Primary immunodeficiency,
GATA2 deficiency. Calcified hilar lymph node located in the left lung.
Calcified lesion and soft tissue lesion located in the right lung”.
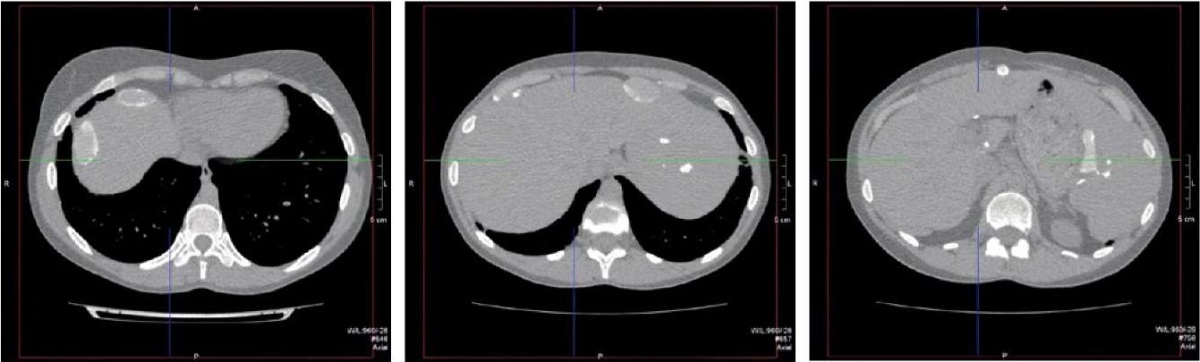
Рисунок 7. Пациентка Ш., 31 год, диагноз —
«Первичное иммунодефицитное состояние, дефицит GATA2.
Обызвествленные лимфатические узлы и очаги
в печени и селезёнке — последствия перенесённого внутрибрюшного туберкулёза».
Figure 7. Patient Sh., 31 years old, diagnosis — “Primary immunodeficiency,
GATA2 deficiency. Calcified lymph nodes and lesions located
in the liver and spleen as the outcome of abdominal tuberculosis”.
В отделении инициирована заместительная терапия: иммуноглобулин человека нормальный для внутривенного введения (ВВИГ) из расчёта 0.4 г/кг массы тела, рекомендовано введение ВВИГ с интервалом 1 раз в 4 недели с учётом отягощённого инфекционного анамнеза, высокий риск развития жизнеугрожающих инфекционных осложнений, глубокий дефицит В-клеточного звена иммунной системы. Скорректирован дефицит железа, подобрана местная терапия.
В связи с высоким риском развития злокачественных костномозговых нарушений пациентка направлена в ФГБУ «НМИЦ гематологии» Минздрава России, где проведено дообследование.
Цитологическое исследование мазка костного мозга (миелограмма) показало следующее: бластные клетки — 0,6% (норма — 0,2–1,7), лимфоциты — 2.8% (норма — 4,3–13,7), эритробласты — 1% (норма — 0,2–1,1), плазматические клетки — 0.4% (норма — 0,1–1,8), мегакариоциты — увеличенное количество, суммарно гранулоцитов — 33.6% (норма — 52,8–68,8), общее количество эритрокариоцитов — 62% (14,6–26,6), тучные клетки — 0.4% (0,0–0,5). Пунктат костного мозга — клеточный. Соотношение ростков кроветворения нарушено. Гранулоцитарный росток сужен (33,6%), с преобладанием незрелых форм. В 10–29% гранулоцитов отмечаются признаки дисплазии (гипогрануляция цитоплазмы, единичные нейтрофилы с пельгеризацией ядра). Снижено количество лимфоцитов (2,8%) и моноцитов (0,2%). Эритроидный росток расширен (62%), обнаруживаются отдельные клетки с неровным контуром ядерной мембраны, фрагментацией ядра, базофильной пунктацией (менее 10%). Признаки дисмегакариоцитопоэза отмечены в 10–29% клеток (фрагментация ядра). Макрофаги в редких полях зрения.
Согласно заключению гистологического исследования трепанобиоптата, в трепанобиоптате костного мозга морфологическая картина характеризуется гипоплазией кроветворной ткани с признаками дизэритропоэза и дисмегакариоцитопоэза (с учётом предоставленных данных) при миелодиспластическом синдроме. На основании проведённого обследования выставлен диагноз «Миелодиспластический синдром с мультилинейной дисплазией (низкий риск по IPSS-R), ассоциированный с герминальным дефектом в гене GATA2».
За время наблюдения в отделении и в период обследования в ФГБУ «НМИЦ гематологии» Минздрава России состояние пациентки оставалось стабильным, сохранялась ремиссия инфекционных осложнений. В настоящее время (август 2023 г.) пациентка находится на амбулаторном наблюдении у иммунолога и с учётом выявленного миелодиспластического синдрома с мультилинейной дисплазией рассматривается в качестве кандидата на алло-ТГСК. Вопрос о возможности и целесообразности проведения алло-ТГСК будет решаться консилиумом, после получения результатов обследования единственного сиблинга (сестры) на наличие дефектов гена GATA2.
С учётом клинической картины, анамнеза, результатов проведённого обследования подтвержден диагноз «Первичное иммунодефицитное состояние, дефицит GATA2». Выявленный вариант c.386del в экзоне 3 гена GATA2, приводящий к сдвигу рамки считывания, ранее не был описан в литературе, однако с учётом характерных клинических проявлений с большой вероятностью он приводит к потере функции соответствующей копии гена [14]. Провести функциональные тесты в настоящее время не представляется возможным. Секвенирование трио по методу Сэнгера для подтверждения de novo статуса выявленного генетического дефекта также не проводилось в связи с полным отсутствием у пациентки контакта с родителями. Единственным доступным родственником является сестра (сиблинг, 1990 г. р.), у которой отсутствуют клинические проявления заболевания. В настоящее время сестре проводится молекулярно-генетическое исследование, и она рассматривается в качестве донора для проведения ТГСК.
Отличительной особенностью данного клинического случая является ранний дебют первых проявлений заболевания (в возрасте семи лет), в виде появления простых бородавок на коже кистей, торпидных к местной терапии и хирургическому лечению, а также лимфостаза нижней конечности, неясного генеза. Развитие первой клинически значимой бактериальной инфекции в возрасте 22 лет патогномонично для данного заболевания, однако возбудителем генерализованного инфекционного процесса являлась именно Mycobacterium tuberculosis (по данным представленной медицинской документации), в то время как для дефицита GATA2 более характерны диссеминированные инфекции, вызванные нетуберкулезными микобактериями (чаще всего вызванные Mycobacterium avium complex) [4][20]. Несмотря на агрессивное течение генерализованного веррукоза, тяжёлого течения микобактериальной инфекции, пациентка в течение длительного времени не была консультирована иммунологом, первое иммунологическое обследование было проведено лишь 25 лет спустя с момента дебюта заболевания, несмотря на то что цитопенический синдром, инфекционные проявления и спленомегалия были выявлены в возрасте 15 лет.
Длительное лечение у фтизиатра, периодический характер цитопении, а также редкость данной патологии — наиболее вероятные причины задержки постановки диагноза и направления пациентки к иммунологу.
В настоящее время ведущими симптомами, ухудшающими качество жизни пациентки, являются веррукозные элементы на коже верхних конечностей, и в меньшем количестве — на коже лица и туловища, стоп. По данным мировой литературы, симптоматическая терапия, включающая хирургическое лечение, малоэффективна, хотя описаны случаи эффективности применения препаратов с иммуномодулирующим эффектом у пациентов с рецидивирующим веррукозом [21][22], однако существенного эффекта возможно достичь лишь после проведения ТГСК. С учётом высокого риска ТГКС у пациентов с дефицитом GATA2, отсутствия в настоящее время донора для проведения процедуры инициирована терапия изопринозином пролонгированным курсом совместно с регулярной, рутинной обработкой пораженных участков кожи антисептическими, кератолитическими средствами.
С целью профилактики инфекций при практически полном отсутствии В-лимфоцитов (уровень CD19+ — 0,4 кл/мкл, норма — 100–500 кл/мкл) и, как следствие, снижении уровня IgG до 528 мг/дл (норма — 900–1800), IgA — 69 мг/дл (норма — 100–350) инициирована заместительная терапия ВВИГ в дозе 0.4 г/кг массы тела с интервалом 1 раз в 4 недели. В литературе имеются данные об эффективности использования препаратов ВВИГ в отношении инфекционных осложнений у пациентов с дефицитом GATA2 [23].
Единственным куративным методом лечения данной формы ПИД является ТГС, однако принятие решения о проведении ТГКС у пациентов старше 18 лет представляет серьёзные трудности. Опыт проведения ТГКС у взрослых в мире достаточно мал, в России он исчисляется единичными случаями. Кроме того, прогноз ТГКС, по данным литературы, представляет серьёзные риски. С другой стороны, с учётом крайне низкого качества жизни пациентки, онкопатологии (характерной для данного заболевания — рака вульвы), развития синдрома миелодисплазии, высокого риска развития лейкоза, выраженного желания пациентки пройти данный вид лечения в настоящее время проводится поиск донора для ТГСК.
Дефицит GATA2 является орфанным заболеванием, которое (с учётом широкого спектра клинических проявлений и возраста дебюта) может встречаться в практике врачей различных специальностей, в том числе фтизиатров, дерматологов, сосудистых хирургов, гинекологов, отоларингологов и педиатров. Диагностику данного заболевания также затрудняют симптомы не характерные для ПИД: нейросенсорная тугоухость, лимфостаз, рак вульвы. Описанный нами клинический случай подчеркивает важность повышения настороженности врачей смежных специальностей в отношении первичных иммунодефицитных состояний для более ранней постановки диагноза и инициации адекватной терапии. Кроме того, следует отметить ценность совместного ведения пациентов с ПИД иммунологами и гематологами. Данный клинический пример демонстрирует своевременное проведение трепанобиопсии костного мозга и выявление миелодиспластического синдрома на ранней стадии риска. В статье представлено подробное описание клинической картины и особенностей течения первичного иммунодефицитного состояния, вызванного ранее не описанным в литературе вариантом c.386del в гене GATA2 у взрослой пациентки.
1. Bousfiha A, Moundir A, Tangye SG, Picard C, Jeddane L, et al. The 2022 Update of IUIS Phenotypical Classification for Human Inborn Errors of Immunity. J Clin Immunol. 2022;42(7):1508-1520. https://doi.org/10.1007/s10875-022-01352-z
2. Сизякина Л.П., Андреева И.И. Справочник по клинической иммунологии. Ростов-на-Дону: Феникс; 2005.
3. Кондратенко И.В., Бологов А.А. Первичные иммунодефициты: учебное пособие. Москва: ИндексМед Медиа; 2020.
4. Vinh DC, Patel SY, Uzel G, Anderson VL, Freeman AF, et al. Autosomal dominant and sporadic monocytopenia with susceptibility to mycobacteria, fungi, papillomaviruses, and myelodysplasia. Blood. 2010;115(8):1519-29. https://doi.org/10.1182/blood-2009-03-208629
5. Bigley V, Haniffa M, Doulatov S, Wang XN, Dickinson R, et al. The human syndrome of dendritic cell, monocyte, B and NK lymphoid deficiency. J Exp Med. 2011;208(2):227-34. https://doi.org/10.1084/jem.20101459
6. Mansour S, Connell F, Steward C, Ostergaard P, Brice G, et al. Emberger syndrome-primary lymphedema with myelodysplasia: report of seven new cases. Am J Med Genet A. 2010;152A(9):2287-96. https://doi.org/10.1002/ajmg.a.33445
7. Hahn CN, Chong CE, Carmichael CL, Wilkins EJ, Brautigan PJ, et al. Heritable GATA2 mutations associated with familial myelodysplastic syndrome and acute myeloid leukemia. Nat Genet. 2011;43(10):1012-7. https://doi.org/10.1038/ng.913
8. Hsu AP, Sampaio EP, Khan J, Calvo KR, Lemieux JE, et al. Mutations in GATA2 are associated with the autosomal dominant and sporadic monocytopenia and mycobacterial infection (MonoMAC) syndrome. Blood. 2011;118(10):2653-5. https://doi.org/10.1182/blood-2011-05-356352
9. Rodrigues NP, Tipping AJ, Wang Z, Enver T. GATA-2 mediated regulation of normal hematopoietic stem/progenitor cell function, myelodysplasia and myeloid leukemia. Int J Biochem Cell Biol. 2012;44(3):457-60. https://doi.org/10.1016/j.biocel.2011.12.004
10. Li Y, Qi X, Liu B, Huang H. The STAT5-GATA2 pathway is critical in basophil and mast cell differentiation and maintenance. J Immunol. 2015;194(9):4328-38. https://doi.org/10.4049/jimmunol.1500018
11. Homan CC, Venugopal P, Arts P, Shahrin NH, Feurstein S, et al. GATA2 deficiency syndrome: A decade of discovery. Hum Mutat. 2021;42(11):1399-1421. https://doi.org/10.1002/humu.24271
12. Kozyra EJ, Pastor VB, Lefkopoulos S, Sahoo SS, Busch H, et al. Synonymous GATA2 mutations result in selective loss of mutated RNA and are common in patients with GATA2 deficiency. Leukemia. 2020;34(10):2673-2687. https://doi.org/10.1038/s41375-020-0899-5
13. Oleaga-Quintas C, de Oliveira-Júnior EB, Rosain J, Rapaport F, Deswarte C, et al. Inherited GATA2 Deficiency Is Dominant by Haploinsufficiency and Displays Incomplete Clinical Penetrance. J Clin Immunol. 2021;41(3):639-657. https://doi.org/10.1007/s10875-020-00930-3
14. Spinner MA, Sanchez LA, Hsu AP, Shaw PA, Zerbe CS, et al. GATA2 deficiency: a protean disorder of hematopoiesis, lymphatics, and immunity. Blood. 2014;123(6):809-21. https://doi.org/10.1182/blood-2013-07-515528
15. Donadieu J, Lamant M, Fieschi C, de Fontbrune FS, Caye A, et al. Natural history of GATA2 deficiency in a survey of 79 French and Belgian patients. Haematologica. 2018;103(8):1278-1287. https://doi.org/10.3324/haematol.2017.181909
16. Mardahl M, Jørgensen SE, Schneider A, Raaschou-Jensen K, Holm M, et al. Impaired immune responses to herpesviruses and microbial ligands in patients with MonoMAC. Br J Haematol. 2019;186(3):471-476. https://doi.org/10.1111/bjh.15947
17. Haugas M, Lilleväli K, Hakanen J, Salminen M. Gata2 is required for the development of inner ear semicircular ducts and the surrounding perilymphatic space. Dev Dyn. 2010;239(9):2452-69. https://doi.org/10.1002/dvdy.22373
18. Parta M, Shah NN, Baird K, Rafei H, Calvo KR, et al. Allogeneic Hematopoietic Stem Cell Transplantation for GATA2 Deficiency Using a Busulfan-Based Regimen. Biol Blood Marrow Transplant. 2018;24(6):1250-1259. https://doi.org/10.1016/j.bbmt.2018.01.030
19. Grossman J, Cuellar-Rodriguez J, Gea-Banacloche J, Zerbe C, Calvo K, et al. Nonmyeloablative allogeneic hematopoietic stem cell transplantation for GATA2 deficiency. Biol Blood Marrow Transplant. 2014;20(12):1940-8. https://doi.org/10.1016/j.bbmt.2014.08.004
20. Marciano BE, Olivier KN, Folio LR, Zerbe CS, Hsu AP, et al. Pulmonary Manifestations of GATA2 Deficiency. Chest. 2021;160(4):1350-1359. https://doi.org/10.1016/j.chest.2021.05.046
21. W est ES, Kingsbery MY, Mintz EM, Hsu AP, Holland SM, et al. Generalized verrucosis in a patient with GATA2 deficiency. Br J Dermatol. 2014;170(5):1182-6. https://doi.org/10.1111/bjd.12794
22. Rivera A, Tyring SK. Therapy of cutaneous human Papillomavirus infections. Dermatol Ther. 2004;17(6):441-8. https://doi.org/10.1111/j.1396-0296.2004.04047.x
23. Bogaert DJ, Laureys G, Naesens L, Mazure D, De Bruyne M, et al. GATA2 deficiency and haematopoietic stem cell transplantation: challenges for the clinical practitioner. Br J Haematol. 2020;188(5):768-773. https://doi.org/10.1111/bjh.16247
Евгений Александрович Фролов – врач аллерголог-иммунолог, младший научный сотрудник отделения Иммунопатологии
Москва
Авторы заявляют об отсутствии конфликта интересов
Фатима Ильясовна Абдулаева – врач-ординатор
Москва
Авторы заявляют об отсутствии конфликта интересов
Юлия Алексеевна Горностаева – к.м.н., старший научный сотрудник отделения Иммунопатологии
Москва
Авторы заявляют об отсутствии конфликта интересов
Татьяна Васильевна Латышева – д.м.н., проф., профессор кафедры клинической аллергологии и иммунологии; заведующий отделения интенсивной терапии
Москва
Авторы заявляют об отсутствии конфликта интересов
Елена Александровна Латышева – д.м.н., доцент кафедры Клинической иммунологии факультета; заведующий отделения Иммунопатологии
Москва
Авторы заявляют об отсутствии конфликта интересов
Гулюмхан Эльвировна Аминова – врач-рентгенолог, заведующий отделением лучевой диагностики
Москва
Авторы заявляют об отсутствии конфликта интересов
Фролов Е.А., Абдулаева Ф.И., Горностаева Ю.А., Латышева Т.В., Латышева Е.А., Аминова Г.Э. Особенности клинической картины и течения дефицита GATA2, осложнённого генерализованным веррукозом с исходом в миелодиспластический синдром во взрослом возрасте. Медицинский вестник Юга России. 2023;14(4):35-43. https://doi.org/10.21886/2219-8075-2023-14-4-35-43
Frolov E.A., Abdulaeva F.I., Gornostaeva U.A., Latysheva T.V., Latysheva E.A., Aminova G.E. Features of the clinical picture and course of GATA2 deficiency complicated by generalized verrucosis with an outcome in myelodysplastic syndrome in adulthood. Medical Herald of the South of Russia. 2023;14(4):35-43. (In Russ.) https://doi.org/10.21886/2219-8075-2023-14-4-35-43
344022, г. Ростов-на-Дону, пер. Нахичеванский, 29
Ростовский государственный медицинский университет
Тел.: +7 918 571 0558
E-mail: journal@medicalherald.ru


































