



Contents
Scroll to:
https://doi.org/10.21886/2219-8075-2023-14-4-35-43
Scroll to:
GATA2 deficiency is a rare disease belonging to the group of phagocyte birth defects, which is clinically manifested by four syndromes: MonoMac syndrome (myedysplasia and immunodeficiency associated with the development of infections caused by Mycobacterium avium complex); monocyte, dendritic cell, B- and NK-lymphocyte deficiency syndrome; Emberger syndrome, including primary lymphedema with myelodysplasia and sensorineural hearing loss, as well as familial myelodysplastic syndrome and acute myeloid leukemia. The disease is inherited by autosomal dominant type, but in most cases, mutations ofthe germ line of the GATA2 gene occur de novo. The first manifestations of the disease occur in early adulthood, the course of GATA2 deficiency is variable and may differ in individuals in the same family with similar genetic variants. The article presents a clinical case of manifestation of GATA2 deficiency at the age of seven years in the form of development of generalized verrucosis, lymphostasis of the lower limb, generalized tuberculosis with involvement of the abdominal cavity, small pelvis, and chest organs. The examination revealed deficiency of monocytes, B- and NK-lymphocytes, myelodysplastic syndrome with multilineage dysplasia. We present a detailed description of the clinical picture and peculiarities of the course of the primary immunodeficiency state, the results of the examination and treatment.
Frolov E.A., Abdulaeva F.I., Gornostaeva U.A., Latysheva T.V., Latysheva E.A., Aminova G.E. Features of the clinical picture and course of GATA2 deficiency complicated by generalized verrucosis with an outcome in myelodysplastic syndrome in adulthood. Medical Herald of the South of Russia. 2023;14(4):35-43. (In Russ.) https://doi.org/10.21886/2219-8075-2023-14-4-35-43
Currently, more than 480 different forms of congenital errors in the immune system, specified as primary immunodeficiencies (PID), are presented in world literature; they are divided into 10 groups depending on the affected part of the immune system [1]. The clinical presentation of PID is variable and characterized by increased susceptibility to infections, as well as by autoimmune and autoinflammatory syndromes, pathological lymphoproliferation, bone marrow failure, and/or oncological diseases [2][3]. One of the representatives of PID conditions combining a variety of clinical manifestations is GATA2 deficiency, which is included in the group of congenital defects in the amount or functions of phagocytes according to the international classification.
The history of the discovery of GATA2 deficiency dates back to 2010, when a group of scientists from the USA [4] first described the syndrome of immunodeficiency and myelodysplasia, manifested by a profound deficiency of monocytes in the peripheral blood and a tendency to develop infections caused by the Mycobacterium avium complex; it was abbreviated as MonoMAC. At the same time, three more similar syndromes were described including the syndrome of deficiency in monocyte, dendritic cells, B and NK lymphocytes; Emberger syndrome with primary lymphedema, myelodysplasia, and sensorineural hearing loss; and familial myelodysplastic syndrome (MDS) with acute myeloid leukemia [5–7]. In 2011, it was revealed that all four syndromes resulted from germline mutations in the GATA2 gene and came out in different manifestations of the same disease, subsequently named GATA2 deficiency.
The GATA2 gene is located on the long arm of the third chromosome; the inheritance mechanism of this disease is autosomal dominant [8]. The GATA2 protein belongs to a family of nuclear transcription factors that bind to DNA consensus sequences and other transcription factors, regulating the expression of multiple target genes [9]. In humans, it is expressed in hematological cells at the stem cell stage and later stages during progenitor cell development. Increased and/or decreased gene expression regulates the progression of these immature cells into their final forms, as well as into certain types of tissue cells such as macrophages and mast cells [10].
To date, about 500 patients with GATA2 deficiency have been reported worldwide [11] and more than 150 different pathogenic variants of the gene have been described, which arise de novo in most cases [12]. The course of GATA2 deficiency is variable and may differ among individuals within the same family who have similar genetic variants. Most often, the first manifestations of the disease occur in early adulthood. The disease phenotype is heterogeneous; it does not have a clear correlation with the genotype and is characterized by incomplete clinical penetrance [13]. Symptoms may incorporate severe recurrent infections including viral, bacterial, and fungal ones; cytopenias; lymphedema; alveolar proteinosis and malignant myeloid diseases [14][15]. Infectious complications due to GATA2 deficiency are one of the main causes of disability in these patients. They are associated with deficiencies of B lymphocytes, NK cells, and defects in the congenital immune response including impaired production of type I interferon [16], and are characterized by increased susceptibility to viral infections such as infections caused by human papillomavirus (HPV) and simplex herpes, non-tuberculous mycobacteria, and other more common bacterial respiratory infections. Hearing loss is also diagnosed as a part of the symptom complex of GATA2 deficiency; it is caused by the participation of the gene in the vestibular morphogenesis of the semicircular canals and the formation of the perilymphatic space around the semicircular canals of the inner ear [17].
Currently, the only curative treatment for GATA2 deficiency is allogeneic hematopoietic stem cell transplantation (HSCT). As in most cases of PID, resolving the issue of HSCT is a serious task for oncologists and hematologists and requires a comprehensive assessment of the patient’s current condition. Foreign scientific literature provides data on a 86% survival rate for two years after HSCT (n = 22, average age is 26 years) [18], 57% survival rate for 3.5 years after HSCT (n = 14, average age is 33 years ) [19], 72%, 65%, and 54% survival rates for 1, 2 and 4 years after HSCT, respectively (n = 21, patients aged 15 to 48 years) [14]; however, these cohorts cannot be compared between themselves due to differences in the severity of clinical manifestations of the disease and conditioning regimens.
Patient Sh. is 31 years old. She was admitted to the department with complaints of unstable stool with a tendency to diarrhea, burning and pain when urinating, and recurrent warts.
As a child, she grew and developed according to age standards. There is no family history but the maternal grandfather complained of significant hearing loss. In the period from 3 to 18 years, frequent mild exacerbations of otitis occurred after acute respiratory viral infections (up to four times a year), treatment was local and symptomatic with a positive effect.
Since the age of 7, simple warts started arising on the skin of the hands for the first time, and since the age of 24 (2016), condylomas in the genital area emerged. The warts increased in size and number with age (Figures 1, 2). Repeated courses of external therapy and systemic immunomodulatory therapy were carried out without visible improvement. Removal of warts by cryodestruction was ineffective, however, in places where the radio wave method of removal was used, new elements did not emerge, but keloid scars subsequently remained.

Figure 1. Patient Sh., 31 years old, diagnosis — “Primary immunodeficiency,
GATA2 deficiency. Generalized verrucosis, predominantly acral localization”.
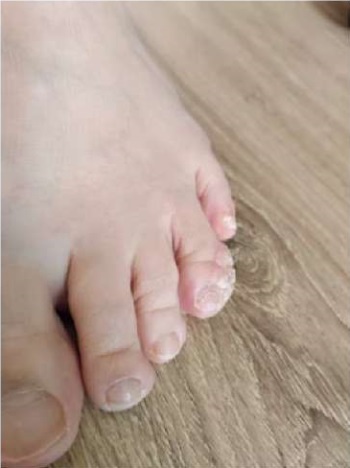
Figure 2. Patient Sh., 31 years old, diagnosis — “Primary immunodeficiency,
GATA2 deficiency. Vulgar plantar warts”.
Since the age of 11, lymphostasis of the right lower limb of unspecified origin developed and started progressing; at the age of 20, an operation was performed to form a lymphovenous anastomosis without significant effect (Figure 3).
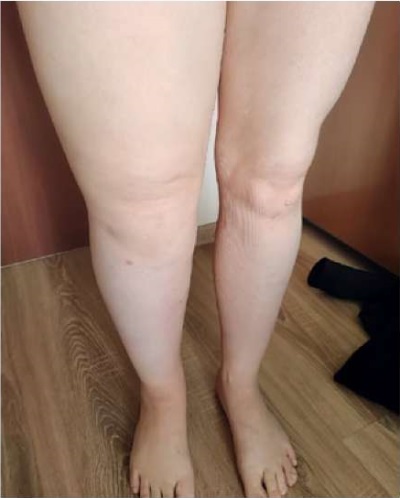
Figure 3. Patient Sh., 31 years old, diagnosis — “Primary immunodeficiency,
GATA2 deficiency. Lymphostasis of the right lower limb”.
At the age of 15, generalized tuberculosis affecting the abdominal cavity, pelvis, and chest organs was diagnosed. Mycobacterium tuberculosis was detected during a bacteriological examination of pleural fluid, as well as materials discharged from the pelvic abscess and from small intestinal fistulas. For 2 years, inpatient conservative therapy and repeated surgical interventions were carried out including resection of the transverse colon, resection of the ascending colon, right tubectomy for pelvic tuberculosis (laparotomy), opening and drainage of a pelvic abscess (Figure 4). In the period from 15 to 30 years old, she was registered at an anti-tuberculosis dispensary, and the Diaskintest result remained negative. Since December 2022, she has been deregistered, and clinical and laboratory remission of tuberculosis has been confirmed.
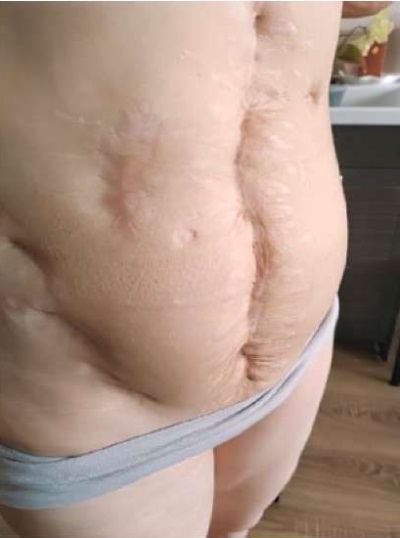
Figure 4. Patient Sh., 31 years old, diagnosis — “Primary immunodeficiency,
GATA2 deficiency. Tuberculous lung lesion, visceral tuberculosis,
genital tuberculosis”. Condition after surgical treatment (2007–2009).
Since the age of 16 (2009), tests revealed anemia of mixed origin, namely, iron deficiency and pernicious ones. A clinical blood test revealed leukopenia (3.8×10^9/l) for the first time.
At the age of 18, a study of the immune status was carried out at KVKD No. 1 and the following indicators were detected: immunoglobulin A (IgA) was 1.7 g/l (norm 0.9–5.0); IgG was 30 g/l (norm 9–20); IgM was 4.9 g/l (norm 0.7–3.7); leukocytes were 3.4x10^9/l, lymphocytes were 0.9x10^9/l, CD3+ T-lymphocytes were 611 cells/μl (norm 800–2200), CD4+ T-lymphocytes were 275 cells/μl (norm 600–1600), CD8+ T-lymphocytes were 321 cells/µl (norm 300–800), CD19+ B-lymphocytes were 19 cells/µl (norm 100–500).
At the age of 22, the patient was assigned to inpatient treatment at the National Research and Clinical Center for Tuberculosis due to complaints of lower back pain. There was no evidence of tuberculosis of the urinary system; the Diaskintest was negative. A bacteriological examination of urine revealed the growth of Proteus mirabilis (10^5). According to the results of a clinical blood test, hemoglobin was 116 g/l, leukocytes were 3.2x109/l, lymphocytes were 0.8x109/l, granulocytes were 2.2x109/l, monocytes were 0.064x109/l, and platelets were 243x109/l. An ultrasound examination of the abdominal organs detected moderate hepatosplenomegaly, as well as calcifications of the liver and spleen.
At the age of 29, the patient suffered a severe COVID-19 viral infection, confirmed by a PCR study of discharge from the throat/nasopharynx; a degree of lung damage was KT-2. The tests showed an increase in C-reactive protein up to 186 mg/dl. The results of a clinical blood test were as follows: hemoglobin was 123 g/l, erythrocytes were 4.53x10^12/l, leukocytes were 3.73x10^9/l, neutrophils were 3.07x10^9/l, lymphocytes were 0.24x10^9/l, and platelets were 179×10^9/l.
At the age of 30 (2023), the patient was sent for inpatient treatment to the gynecological oncology department of Clinical Hospital No. 4. The endometrial polyp, left outer labia, and condylomas of the genital area were removed. Histological examination revealed vulvar carcinoma in situ (T0-1N0M0). An MRI of the pelvic organs was performed, and enlarged lymph nodes and focal changes in the pelvic bones were detected but macroscopic signs of tumor lesions of the vulva and vagina were not identified.
At the age of 31, the patient first consulted an immunologist. An examination was performed at the Federal State Budgetary Institution State Scientific Center Institute of Immunology of the Federal Medical and Biological Agency of Russia. The results of a molecular genetic study, namely full exome sequencing, revealed a previously undescribed suspicious pathogenic variant c.386del in a heterozygous state in exon 3 (of 6 exons total) of the GATA2 gene; the variant was in a heterozygous dominant state and led to a shift in the reading frame of p.Ser129Ter.
The patient’s height was 165 cm, body weight was 67 kg. The condition was stable, serious due to the underlying disease. Consciousness was clear. Skin was with the following features: multiple verrucous elements on the skin of the dorsal and palmar surfaces of the hands, fingers; vulgar warts on the skin of the feet (Figures 4, 5); single verrucous elements on the skin of the face in frontal, nasal areas, and lower extremities. Multiple keloid scars were found in the urogenital area. Scars in the abdominal area and lumbus remained after laparoscopic operations (Fig. 7). Lymphostasis of the right lower limb was revealed (Figure 6). Nasal breathing was free. Breathing was harsh, wheezing could not be heard. Heart sounds were clear, and the rhythm was correct. Hemodynamic parameters were stable.
A clinical blood test revealed anemia: hemoglobin was 85 g/l, erythrocytes were 3.03x10^12/l; leukopenia, lymphopenia (leukocytes were 2.1x10^9/l, neutrophils were 1.7x10^9/l, lymphocytes were 0.31×10^9/l, monocytes were 0.01×10^9/l); platelets were 179×10^9/l; a slight increase in ESR up to 30 mm/h according to Westergren.
A biochemical blood test revealed a decrease in the level of total protein to 50 g/l (norm 66–83 g/l); creatinine was 50 µmol/l (norm 58–96 µmol/l), iron was 2.2 µmol/l (norm 10.7–32.2 µmol/l), ferritin was 26 ng/ml (norm 11–306.8 ng/ml), C-reactive protein was 6.6 mg/dl (norm <5 mg/dl).
In the immunological examination, attention was drawn to a decrease in immunoglobulin A (IgA) to 69 mg/dl (norm 100-350), IgG to 528 mg/dl (norm 900–1800) with a normal level of IgM of 89 mg/dl (norm 80–250). In addition, tests revealed a sharp decrease in CD3+ T cells to 177 cells/μl (norm 800–2200), in CD3+CD4+ T helper cells to 68 cells/μl (norm 600–1600), in CD3+CD8+ T-cytotoxic cells to 97 cells/µl (norm 190–650), in CD3‒CD16,56+ NK cells to 115 cells/µl (norm 150–600), in CD19+ B-cells to 0.4 cells/µl (norm 100–500) (Table 1). Given the low amount of B lymphocytes in the peripheral blood, phenotyping of B lymphocytes was not performed. A study of phagocytic activity was not carried out due to the lack of technical capabilities during the patient’s hospitalization; it will be carried out in the future.
Table 1
Results of immunophenotyping of peripheral blood lymphocytes of the patient
|
Indicators |
Result |
Normal |
|
Lymphocytes (per 1 μl of blood) |
300 |
1200–3000 |
|
CD3+ T-cells (% lymphocytes) |
59.0 |
55–80 |
|
CD3+ T-cells (per 1 μl of blood) |
177 |
800–2200 |
|
CD3+CD4+ T-helpers (% lymphocytes) |
22.6 |
31–49 |
|
CD3+CD4+ T-helpers (per 1 μl of blood) |
68 |
600–1600 |
|
CD3+CD8+ cytotoxic T-cells (% lymphocytes) |
32.3 |
12–30 |
|
CD3+CD8+ cytotoxic T-cells (per 1 μl of blood) |
97 |
190–650 |
|
CD3+CD8+ |
0.70 |
1.5–3.0 |
|
CD3+CD4+CD8+ (% lymphocytes) |
1.6 |
< 2 |
|
CD3‒CD16,56+ NK-cells (% lymphocytes) |
38.5 |
6–20 |
|
CD3‒CD16,56+ NK-cells (per 1 μl of blood) |
115 |
150–600 |
|
CD19+ B-cells (% lymphocytes) |
0.14 |
5–19 |
|
CD19+ B-cells (per 1 μl of blood) |
0.4 |
100–500 |
According to a PCR blood test, DNA of cytomegalovirus, Epstein-Barr virus, and herpes virus type 6 were not detected. A PCR study of scrapings from verrucous elements detected 5.9 copies of DNA of HPV type 16 per 10^5 cells.
According to a PCR study of scrapings from the mucous membrane in the genital area (in the amount of HPV DNA copies per 10^5 cells), HPV35 (7.8), HPV16 (8.0), HPV51 (7.9) were detected.
Ultrasound of regional lymph nodes revealed no data on pathologically changed lymph nodes.
According to abdominal ultrasound, calcifications were detected in the 4th, 6th, and 7th segments of the liver measuring 1.0 cm, 3.5 cm, and 0.9 cm, respectively; and moderate splenomegaly was also detected (length 143 mm, thickness 48 mm).
The conclusion of the CT scan of the chest was as follows: post-tuberculosis, fibrous, post-inflammatory changes in the lungs; residual manifestations of interstitial, probably, viral pneumonia with signs of interstitial fibrosis; multiple lesion foci in the lungs; a new lesion focus in S10 of the lower lobe of the right lung; compaction (nodular formation?) in the cortical parts of S10 of the lower lobe of the right lung (Figures 5, 6, 7).
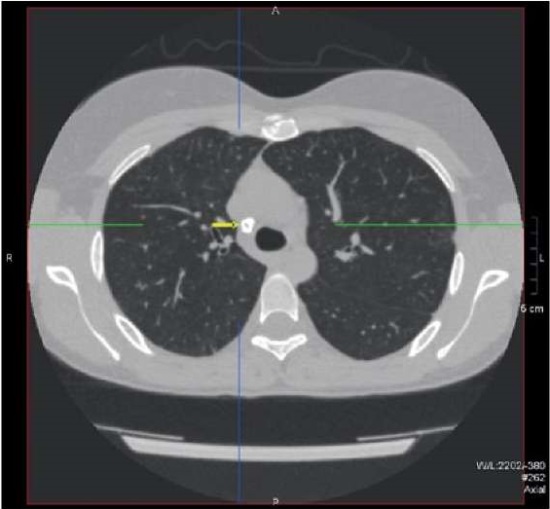
Figure 5. Patient Sh., 31 years old, diagnosis — “Primary immunodeficiency,
GATA2 deficiency. Calcified paratracheal lymph node”.
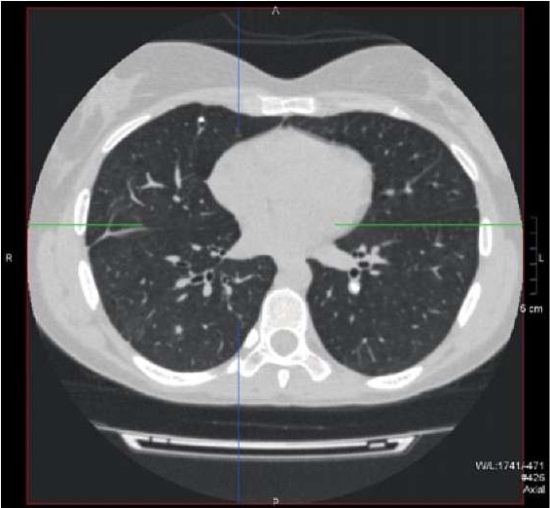
Figure 6. Patient Sh., 31 years old, diagnosis — “Primary immunodeficiency,
GATA2 deficiency. Calcified hilar lymph node located in the left lung.
Calcified lesion and soft tissue lesion located in the right lung”.
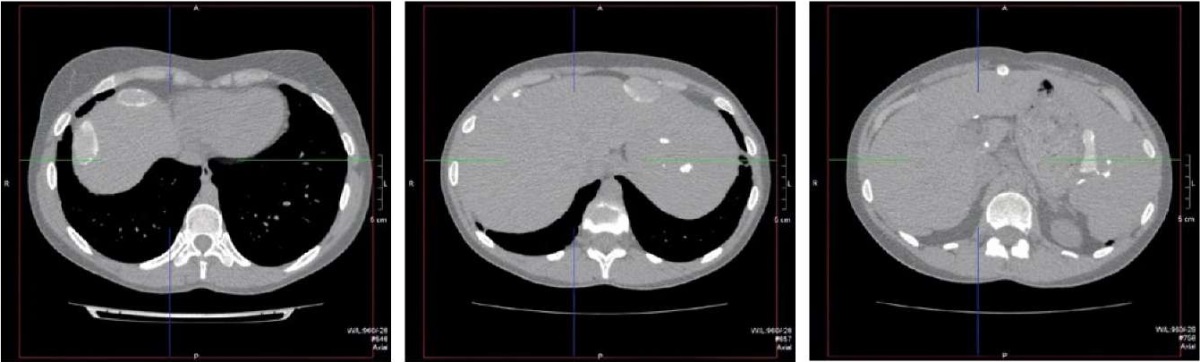
Figure 7. Patient Sh., 31 years old, diagnosis — “Primary immunodeficiency,
GATA2 deficiency. Calcified lymph nodes and lesions located in the liver
and spleen as the outcome of abdominal tuberculosis”.
Replacement therapy was initiated in the department as an infusion of normal human immunoglobulin for intravenous administration (IVIG) in an amount of 0.4 g/kg body weight. IVIG administration was recommended at intervals of once every 4 weeks taking into account a burdened infectious history, a high risk of developing life-threatening infectious complications, and a deep deficiency of the B-cellular part of the immune system. As a result, iron deficiency was corrected and local therapy was selected.
Due to the high risk of developing malignant bone marrow disorders, the patient was sent to the Federal State Budgetary Institution “National Medical Research Center of Hematology” of the Ministry of Health of Russia, where further examination was carried out.
A cytological examination of a bone marrow smear (myelogram) showed the following: blast cells were 0.6% (norm 0.2–1.7), lymphocytes were 2.8% (norm 4.3–13.7), erythroblasts were 1% (norm 0.2–1.1), plasma cells were 0.4% (norm 0.1–1.8), megakaryocytes were increased in number, total granulocytes were 33.6% (norm 52.8–68.8), the total number of erythrokaryocytes was 62% (14.6–26.6), mast cells were 0.4% (0.0–0.5). Bone marrow punctate was cellular. The ratio of hematopoietic lineages was disturbed. The granulocytic lineage was narrowed (33.6%), with a predominance of immature forms. Signs of dysplasia such as hypogranulation of the cytoplasm and emergence of single neutrophils with pelgerization of the nucleus were revealed in 10–29% of granulocytes. The amount of lymphocytes (2.8%) and monocytes (0.2%) was reduced. The erythroid lineage was expanded (62%); there were found individual cells (less than 10%) with an uneven contour of the nuclear membrane, fragmentation of the nucleus, and basophilic punctuation. Signs of dysmegakaryocytopoiesis, such as nuclear fragmentation, were noted in 10–29% of cells. Macrophages were seen in the rare viewing area.
The histological examination revealed the morphological picture in the bone marrow trepan-biopsy material, which was characterized by hypoplasia of hematopoietic tissue with signs of dyserythropoiesis and dysmegakaryocytopoiesis, considering the data earlier provided, as at myelodysplastic syndrome. Based on the examination, a diagnosis of “Myelodysplastic syndrome with multilineage dysplasia (low risk according to IPSS-R) associated with a germinal defect in the GATA2 gene” was made.
During observation in the department and during the examination at the Federal State Budgetary Institution “National Medical Research Center for Hematology” of the Ministry of Health of Russia, the patient’s condition remained stable, and remission of infectious complications remained. Currently (August 2023), the patient is under outpatient observation with an immunologist and is being considered as a candidate for allo-HSCT on the base of the identified myelodysplastic syndrome with multilineage dysplasia. The question of the possibility and advisability of allo-HSCT will be decided by a council after receiving the results of examining the only sibling (sister) for the presence of defects in the GATA2 gene.
Considering the clinical picture, anamnesis, and the results of the examination, the diagnosis of “Primary immunodeficiency state, GATA2 deficiency” was confirmed. The identified variant c.386del in exon 3 of the GATA2 gene leading to a reading frameshift has not previously been described in the literature but it seems to cause the function loss of the corresponding copy of the gene [14] with regard to the characteristic clinical manifestations. Currently, it seems to be impossible to perform functional tests. Sequencing of the trio using the Sanger method to confirm the de novo status of the identified genetic defect was also not performed due to the patient’s complete lack of contact with her parents. The only available relative is a sister (sibling, born in 1990), who has no clinical manifestations of the disease. The sister is currently undergoing molecular genetic testing and is being considered as a donor for HSCT.
A distinctive feature of this clinical case is the early debut, at the age of seven years, for the first manifestations of the disease in the form of the emerging simple warts on the skin of the hands, which were torpid to local therapy and surgical treatment, as well as in the form of lymphostasis of the lower limb of unknown origin. The development of the first clinically significant bacterial infection at the age of 22 years was pathognomonic for this disease, however, the causative agent of the generalized infectious process was Mycobacterium tuberculosis according to the presented medical documentation; while for GATA2 deficiency the disseminated infections induced by non-tuberculous mycobacteria (most often by the Mycobacterium avium complex) are more typical [4][20]. Despite the aggressive course of generalized verrucosis and a severe course of mycobacterial infection, the patient was not consulted by an immunologist for a long time. The first immunological examination was carried out only 25 years after the onset of the disease even as the cytopenic syndrome, infectious manifestations and splenomegaly were identified at the age of 15 years.
Long-term treatment by a phthisiatrician, the periodic nature of cytopenia, as well as the rarity of this pathology are the most likely reasons for the delay in diagnosis and referral of the patient to an immunologist.
Currently, the leading symptoms that worsen the patient’s quality of life are verrucous elements on the skin of the upper extremities and, to a lesser extent, on the skin of the face, torso, and feet. According to world scientific literature, symptomatic therapy including surgical treatment is ineffective, although efficacy cases of the using drugs with an immunomodulatory effect in patients with recurrent verrucosis have been described [21][22]. However, a significant effect in these instances can be achieved only after HSCT. Considering the high risk of HSCT in patients with GATA2 deficiency and the current lack of a donor for the procedure, a prolonged course of isoprinosine therapy was initiated together with regular, routine treatment of the affected skin areas with antiseptic and keratolytic agents.
In order to prevent infections at the almost complete absence of B-lymphocytes (CD19+ level is 0.4 cells/μl at norm 100–500 cells/μl) and, as a result, a decrease in the levels of IgG to 528 mg/dl (norm 900–1800) and IgA to 69 mg/dl (norm 100–350), the IVIG replacement therapy was initiated at a dose of 0.4 g/kg body weight at interval of once every 4 weeks. There is data in the literature on the efficacy of using IVIG drugs in relation to infectious complications in patients with GATA2 deficiency [23].
The only curative treatment method for this form of PID is HSCT; however, making a decision to perform HSCT in patients over 18 years of age presents serious difficulties. The world experience of performing HSCT in adults is insufficient; in Russia it is represented by isolated cases. In addition, the prognosis of HSCT, according to the available scientific data, is of serious risk. On the other hand, there is a reason for searching for a donor for HSCT against the background of the extremely low quality of the patient’s life, and factors including diagnosed oncopathology, namely vulvar cancer that is specifically attributed to this disease, the development of myelodysplasia syndrome, the high risk of developing leukemia, and the patient’s expressed desire to undergo this type of treatment. Searching for a donor for HSCT is currently underway.
GATA2 deficiency is an orphan disease; due to the wide range of clinical manifestations and the age of onset, it can be encountered in the practice of doctors of various specialties, including phthisiatricians, dermatologists, vascular surgeons, gynecologists, otolaryngologists, and pediatricians. Diagnosis of this disease is complicated by symptoms not typical of PID such as sensorineural hearing loss, lymphostasis, and vulvar cancer. The clinical case described here emphasizes the importance of increasing the alertness of doctors of related specialties regarding PID conditions for earlier diagnosis and initiation of adequate therapy. In addition, it should be noted the value of joint management of patients with PID on the part of immunologists and hematologists. This clinical example demonstrates the opportuneness of bone marrow trepanobiopsy and identification of myelodysplastic syndrome at an early risk stage. This paper provides a detailed description of the clinical picture and features of the course of a PID state in an adult patient caused by the c.386del variant in the GATA2 gene, previously not described in the literature.
1. Bousfiha A, Moundir A, Tangye SG, Picard C, Jeddane L, et al. The 2022 Update of IUIS Phenotypical Classification for Human Inborn Errors of Immunity. J Clin Immunol. 2022;42(7):1508-1520. https://doi.org/10.1007/s10875-022-01352-z
2. Sizyakina L.P., Andreeva I.I. Spravochnik po klinicheskoj immunologii. Rosotov-on-Don: Feniks; 2005. (In Russ.).
3. Kondratenko I.V., Bologov A.A. Pervichnye immunodeficity: uchebnoe posobie. Moskva: IndeksMed Media; 2020. (In Russ.).
4. Vinh DC, Patel SY, Uzel G, Anderson VL, Freeman AF, et al. Autosomal dominant and sporadic monocytopenia with susceptibility to mycobacteria, fungi, papillomaviruses, and myelodysplasia. Blood. 2010;115(8):1519-29. https://doi.org/10.1182/blood-2009-03-208629
5. Bigley V, Haniffa M, Doulatov S, Wang XN, Dickinson R, et al. The human syndrome of dendritic cell, monocyte, B and NK lymphoid deficiency. J Exp Med. 2011;208(2):227-34. https://doi.org/10.1084/jem.20101459
6. Mansour S, Connell F, Steward C, Ostergaard P, Brice G, et al. Emberger syndrome-primary lymphedema with myelodysplasia: report of seven new cases. Am J Med Genet A. 2010;152A(9):2287-96. https://doi.org/10.1002/ajmg.a.33445
7. Hahn CN, Chong CE, Carmichael CL, Wilkins EJ, Brautigan PJ, et al. Heritable GATA2 mutations associated with familial myelodysplastic syndrome and acute myeloid leukemia. Nat Genet. 2011;43(10):1012-7. https://doi.org/10.1038/ng.913
8. Hsu AP, Sampaio EP, Khan J, Calvo KR, Lemieux JE, et al. Mutations in GATA2 are associated with the autosomal dominant and sporadic monocytopenia and mycobacterial infection (MonoMAC) syndrome. Blood. 2011;118(10):2653-5. https://doi.org/10.1182/blood-2011-05-356352
9. Rodrigues NP, Tipping AJ, Wang Z, Enver T. GATA-2 mediated regulation of normal hematopoietic stem/progenitor cell function, myelodysplasia and myeloid leukemia. Int J Biochem Cell Biol. 2012;44(3):457-60. https://doi.org/10.1016/j.biocel.2011.12.004
10. Li Y, Qi X, Liu B, Huang H. The STAT5-GATA2 pathway is critical in basophil and mast cell differentiation and maintenance. J Immunol. 2015;194(9):4328-38. https://doi.org/10.4049/jimmunol.1500018
11. Homan CC, Venugopal P, Arts P, Shahrin NH, Feurstein S, et al. GATA2 deficiency syndrome: A decade of discovery. Hum Mutat. 2021;42(11):1399-1421. https://doi.org/10.1002/humu.24271
12. Kozyra EJ, Pastor VB, Lefkopoulos S, Sahoo SS, Busch H, et al. Synonymous GATA2 mutations result in selective loss of mutated RNA and are common in patients with GATA2 deficiency. Leukemia. 2020;34(10):2673-2687. https://doi.org/10.1038/s41375-020-0899-5
13. Oleaga-Quintas C, de Oliveira-Júnior EB, Rosain J, Rapaport F, Deswarte C, et al. Inherited GATA2 Deficiency Is Dominant by Haploinsufficiency and Displays Incomplete Clinical Penetrance. J Clin Immunol. 2021;41(3):639-657. https://doi.org/10.1007/s10875-020-00930-3
14. Spinner MA, Sanchez LA, Hsu AP, Shaw PA, Zerbe CS, et al. GATA2 deficiency: a protean disorder of hematopoiesis, lymphatics, and immunity. Blood. 2014;123(6):809-21. https://doi.org/10.1182/blood-2013-07-515528
15. Donadieu J, Lamant M, Fieschi C, de Fontbrune FS, Caye A, et al. Natural history of GATA2 deficiency in a survey of 79 French and Belgian patients. Haematologica. 2018;103(8):1278-1287. https://doi.org/10.3324/haematol.2017.181909
16. Mardahl M, Jørgensen SE, Schneider A, Raaschou-Jensen K, Holm M, et al. Impaired immune responses to herpesviruses and microbial ligands in patients with MonoMAC. Br J Haematol. 2019;186(3):471-476. https://doi.org/10.1111/bjh.15947
17. Haugas M, Lilleväli K, Hakanen J, Salminen M. Gata2 is required for the development of inner ear semicircular ducts and the surrounding perilymphatic space. Dev Dyn. 2010;239(9):2452-69. https://doi.org/10.1002/dvdy.22373
18. Parta M, Shah NN, Baird K, Rafei H, Calvo KR, et al. Allogeneic Hematopoietic Stem Cell Transplantation for GATA2 Deficiency Using a Busulfan-Based Regimen. Biol Blood Marrow Transplant. 2018;24(6):1250-1259. https://doi.org/10.1016/j.bbmt.2018.01.030
19. Grossman J, Cuellar-Rodriguez J, Gea-Banacloche J, Zerbe C, Calvo K, et al. Nonmyeloablative allogeneic hematopoietic stem cell transplantation for GATA2 deficiency. Biol Blood Marrow Transplant. 2014;20(12):1940-8. https://doi.org/10.1016/j.bbmt.2014.08.004
20. Marciano BE, Olivier KN, Folio LR, Zerbe CS, Hsu AP, et al. Pulmonary Manifestations of GATA2 Deficiency. Chest. 2021;160(4):1350-1359. https://doi.org/10.1016/j.chest.2021.05.046
21. W est ES, Kingsbery MY, Mintz EM, Hsu AP, Holland SM, et al. Generalized verrucosis in a patient with GATA2 deficiency. Br J Dermatol. 2014;170(5):1182-6. https://doi.org/10.1111/bjd.12794
22. Rivera A, Tyring SK. Therapy of cutaneous human Papillomavirus infections. Dermatol Ther. 2004;17(6):441-8. https://doi.org/10.1111/j.1396-0296.2004.04047.x
23. Bogaert DJ, Laureys G, Naesens L, Mazure D, De Bruyne M, et al. GATA2 deficiency and haematopoietic stem cell transplantation: challenges for the clinical practitioner. Br J Haematol. 2020;188(5):768-773. https://doi.org/10.1111/bjh.16247
Evgeny A. Frolov – allergist-immunologist, Junior Researcher at the Immunopathology department
Moscow
Authors declares no conflict of interest
Fatima I. Abdulaeva – Resident Physician
Moscow
Authors declares no conflict of interest
Yulia A. Gornostaeva – Cand. Sci. (Med.), Senior Researcher at the Immunopathology department
Moscow
Authors declares no conflict of interest
Tatiana V. Latysheva – Professor, Dr. Sci. (Med.), Professor of the department of Clinical Allergology and Immunology; head of the Intensive Care Unit
Moscow
Authors declares no conflict of interest
Elena A. Latysheva – Dr. Sci. (Med.), Associate Professor of the Department of Clinical Immunology; Head of the Immunopathology Department
Moscow
Authors declares no conflict of interest
Gulyumkhan E. Aminova – radiologist, head of the radiology
department
Moscow
Authors declares no conflict of interest
Frolov E.A., Abdulaeva F.I., Gornostaeva U.A., Latysheva T.V., Latysheva E.A., Aminova G.E. Features of the clinical picture and course of GATA2 deficiency complicated by generalized verrucosis with an outcome in myelodysplastic syndrome in adulthood. Medical Herald of the South of Russia. 2023;14(4):35-43. (In Russ.) https://doi.org/10.21886/2219-8075-2023-14-4-35-43
29, Nakhichevansky Lane, Rostov-on-Don, 344002
Rostov State Medical University
Тel.: +79185710558
e-mail: journal@medicalherald.ru


































