



Содержание
Перейти к:
https://doi.org/10.21886/2219-8075-2022-13-2-141-145
Перейти к:
Цель: представить клинический случай редкого аутовоспалительного заболевания.
Материал и методы: проведен анализ клинического случая синдрома дефицита мевалонаткиназы у девочки 8 лет.
Результаты: синдром дефицита мевалонаткиназы (СДМК) — редкое аутовоспалительное заболевание с аутосомно-рецессивным механизмом наследования. Степень тяжести заболевания имеет корреляцию с остаточной активностью фермента — мевалонаткиназы, участвующего в биосинтезе холестерина и изопреноидов, необходимого для превращение мевалоната в конечный продукт. В результате чего в организме накапливается мевалоновая кислота, особенно высокие уровни её обнаруживаются в моче. Выделяют сравнительно легкий фенотип СДМК: синдром гипериммуноглобулинемии D (впервые описан как HIDS в 1984 г.) и тяжелый вариант — мевалоновую ацидурию. В мире описано чуть больше 300 пациентов с СДМК. Заболевание проявляется периодически возникающей лихорадкой, артралгиями, фарингитом, шейным лимфаденитом, уртикарной сыпью, напоминающей крапивницу, реже васкулитоподобными проявлениями в виде петехиально-пурпурных элементов. Диагностика основывается на активности фермента мевалонаткиназы в крови или клетках кожи, биохимическом анализе мочи (высокие цифры мевалоновой кислоты), а также генетическом подтверждении мутации в гене мевалонаткиназы. Основными принципами терапии аутовоспалительных заболеваний является контроль над клиническими симптомами и над воспалением в целом, а также профилактика амилоидоза.
Заключение: при подозрении на аутовоспалительный процесс в организме необходимо проведение генетического исследования для своевременной диагностики и назначения патогенетической терапии для улучшение качества жизни и предупреждения осложнений.
Сомова Т.М. Аутовоспалительное заболевание синдром гипериммуноглобулинемии D. Медицинский вестник Юга России. 2022;13(2):141-145. https://doi.org/10.21886/2219-8075-2022-13-2-141-145
Somova T.M. Autoinflammatory disease syndrome of hyperimmunoglobulinemia D. Medical Herald of the South of Russia. 2022;13(2):141-145. (In Russ.) https://doi.org/10.21886/2219-8075-2022-13-2-141-145
Аутовоспалительные заболевания (АВЗ) в настоящее время интенсивно изучаются. Большое значение для диагностики АВЗ имеет молекулярно-генетическое тестирование пациентов, поскольку основой развития АВЗ являются патологические мутации, обусловливающие нарушения в системе врождённого (антиген-неспецифического) иммунитета и развитие воспаления. Экспертной группой, в состав которой входят 12 немецких и австрийских ученых, разработан диагностический алгоритм и принципы лечения аутовоспалительных процессов [1]
Аутовоспаление рассматривается как часть этиопатогенеза таких распространённых заболеваний, как болезнь Крона, узловатая эритема, саркоидоз, анкилозирующий спондилоартрит, васкулит и синдромы васкулита, синдром активации макрофагов, моногенный васкулит (STING-ассоцциированный васкулит и дефицит аденозиндезаминазы II типа (DADA2)), болезнь Стилла взрослых, системная красная волчанка, системный ювенильный идиопатический артрит, подагрический артрит, псориаз (мутация гена CARD14) [2].
Основные характеристики АВЗ:
В настоящее время известно более 30 генов, мутации в которых приводят к развитию АВЗ, наиболее распространёнными и хорошо изученными являются гены NLRP3, TNFRSF1A и MVK.
Эти гены вызывают развитие следующих основных моногенных АВЗ: криопирин-ассоциированных периодических синдромов (Сryopyrin-аssociated рeriodic syndromes, CAPS); периодического синдрома, ассоциированного с мутацией гена рецептора фактора некроза опухоли (TNF-receptor-associated periodic syndrome, TRAPS); синдрома гипериммуноглобулинемии Д/дефицита мевалонаткиназы (Hyper-immunoglobulinemia D-syndrome, HIDS) [3-7].
Современная классификация аутовоспалительных заболеваний:
I. Наследственная периодическая лихорадка (типичное нарушение функции инфламмасом):
II. Пиогенные заболевания кожи и костей (возможное участие инфламмасом и других механизмов):
III. Гранулематозные заболевания (нарушение активации NFkB и передачи сигналов посредством ИЛ-10):
IV. Идиопатические синдромы + иные нозологические единицы (неясный патогенез, возможно участие инфламмасом):
Снижение активности мевалонаткиназы приводит к дефициту геранилгеранилпирофосфата (GGPP), одного из конечных продуктов мевалоновой кислоты, участвующего в пренилировании внутриклеточных G-белков, и, как следствие, приводит к избыточной активности прокаспазы, синтеза предшественника интерлейкина-1 (ИЛ-1)-бета и его активной формы [5-13].
Более лёгкий вариант дефицита мевалонаткиназы, также известный как синдром гипер-IgD, встречается чаще и характеризуется повторяющимися эпизодами необъяснимой лихорадки без сопутствующей инфекции. Подобные приступы сопровождаются утомляемостью, ознобом, болями в животе, отёком поражённых лимфатических узлов (лимфаденопатия), сыпью, воспалением суставов (артрит) и болью (артралгия). Дополнительные симптомы включают тошноту, диарею, рвоту, головные боли, небольшие язвы во рту и аномальное увеличение печени и селезёнки (гепатоспленомегалия). Сыпь представлена в виде эритематозных пятен и папул. У некоторых людей наблюдается кашель и воспаление задней стенки глотки (фарингит). В результате длительного течения заболевания развиваются такие осложнения, как амилоидоз почек, контрактуры суставов и спайки брюшной полости [14-16].
Цель исследования — представить клиническое наблюдение аутовоспалительного заболеания у пациента.
Проведён анализ клинического случая гипериммуноглобулинемии D (синдрома дефицита мевалонаткиназы) у девочки 8 лет.
Девочка Р., 8 лет (24.11.2013 г.р.) от 4-й беременности на фоне отягощённого акушерского анамнеза (2 выкидыша на раннем сроке), 2-х срочных родов. Масса при рождении — 3660 г, оценка по Апгар — 8/9 баллов. Профилактические прививки по индивидуальному календарю (БЦЖ, V1 против полиомиелита, V1–V2 против вирусного гепатита В). Перенесенные заболевания — ветряная оспа в 1 год 10 мес., ОРВИ, фолликулярная ангина, острый бронхит, острый тонзиллит, инфекционный мононуклеоз. Наследственность по гематологическим, иммунологическим, онкологическим заболеваниям не отягощена.
Больна с двухлетнего возраста, когда после перенесённой ветряной оспы впервые появился приступ лихорадки до 40 градусов, сопровождающийся увеличением нёбных миндалин, появлением на них гнойных налетов и увеличением шейных лимфоузлов (рис. 1). Данный приступ был расценён как инфекционный процесс, купирован антибактериальной терапией и приемом нестероидных противовоспалительных средств (НПВС).
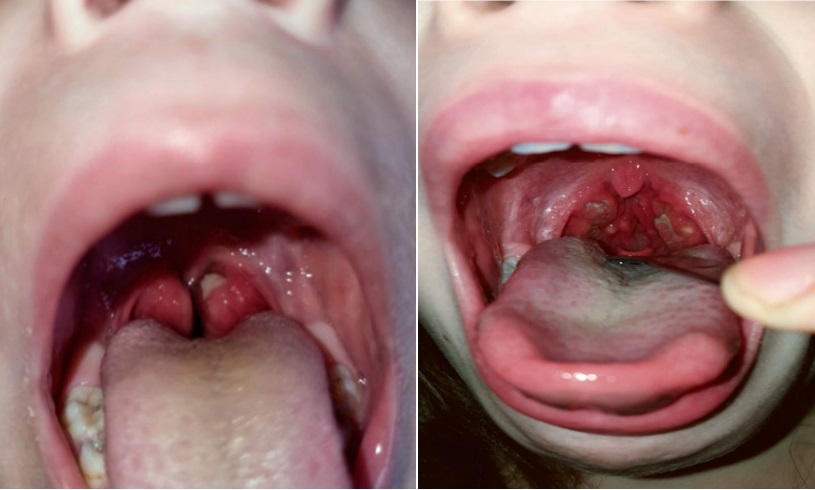
Рисунок 1. Налеты на небных миндалинах в приступный период (собственное наблюдение)
Figure 1. Raids on the palatine tonsils during the onset period (own observation)
Однако далее подобные эпизоды стали рецидивировать каждые 20–30 дней с длительностью приступа 5–7 дней. К температуре и увеличению миндалин стали присоединяться кишечный синдром в виде болей в животе, тошноты, рвоты, жидкого стула; артралгии, а также наблюдалось повышение воспалительной активности крови (СОЭ до 28мм/час).
Вне приступа состояние удовлетворительное, физическое развитие гармоничное по мезосоматическому типу. Интеллект сохранён. По внутренним органам без особенностей. Физиологические оправления в норме.
В течение года отмечался положительный эффект во время приступа на НПВС в виде купирования лихорадки.
С трёхлетнего возраста наблюдалась у иммунолога с диагнозом «Синдром Маршалла», выявлена дисгаммаглобулинемия, в период атак принимала глюкокортикостероиды (ГКС) (преднизолон внутрь 1 мг/кг с положительным эффектом). В возрасте 6-ти лет консультирована ревматологом, проведены иммунологические исследования (экстрагируемые ядерные антитела, антитела к кардиолипину (суммарные), антитела к одно-, двуспиральной ДНК, антитела к циклическим цитруллинированным пептидам, антитела к фосфолипидам), все показатели в пределах нормы. Данных об аутоиммунной патологии нет. В период атак рекомендован приём ГКС (метипред) (1,5 мг/кг). Далее ввиду снижения ответа на ГКС доза преднизолона была постепенно эскалирована до 2 мг/кг в период приступов.
С учётом длительного течения заболевания и коротких интервалов между атаками и ответа только на повышение дозы ГКС до 2 мг/кг в 7,5 лет ребенку было проведено молекулярно-генетическое исследование, в результате которого обнаружена мутация в гене MVK в 11-м экзоне (V377I) и 9-м экзоне (I268T) в гетерозиготном состоянии, что подтверждает диагноз синдрома дефицита мевалонаткиназы (СДМК).
Для определения дальнейшей тактики ведения была направлена в отделение иммунологии ФГБУ «НМИЦ ДГОИ им. Дмитрия Рогачева» Минздрава России, куда была госпитализирована в июле 2021 г.
При поступлении соматический статус стабильный, умеренная гипертрофия миндалин (1–2 степени) с единичными налетами.
Результаты лабораторных тестов:
Результаты инструментальных методов исследования:
Заключение: в исследованном материале признаки активного эрозивного илеита, неактивного реактивного колита. Признаки амилоидоза отсутствуют.
Консультирована офтальмологом — OU без патологических изменений.
Выставлен диагноз «Первичное иммунодефицитное состояние: аутовоспалительный синдром — синдром дефицита мевалонаткиназы (мутация в гене MVK в 11 экзоне (V377I) и 9 экзоне (I268T) в гетерозиготном состоянии)».
Данное зоболевание носит хронический характер, при отсутствии контроля воспалительных приступов высок риск развития амилоидоза почек и полиорганной недостаточности и других несовместимых с жизнью осложнений.
Учитывая тяжесть течения заболевания, а также наличие у пациента рецидивирующих эпизодов высокой лихорадки, сопровождающейся увеличением нёбных миндалин, кишечным синдромом, тошнотой и артралгиями, в рамках достижения и поддержания ремиссии девочке назначен моноклональный ингибитор ИЛ-1b Канакинумаб (Иларис) в дозе 75 мг 1 раз 8 недель, подкожно, по жизненным показаниям.
Уже после первого введения препарата межприступный период увеличился до 50 дней.
Всем детям с синдромом периодической лихорадки показано генетическое исследование для своевременной диагностики редких аутовоспалительных заболеваний, назначения патогенетической терапии для улучшение качества жизни и предупреждения осложнений.
1. Hansmann S, Lainka E, Horneff G, Holzinger D, Rieber N, et al. Consensus protocols for the diagnosis and management of the hereditary autoinflammatory syndromes CAPS, TRAPS and MKD/HIDS: a German PRO-KIND initiative. Pediatr Rheumatol Online J. 2020;18(1):17. DOI: 10.1186/s12969-020-0409-3.
2. Krainer j, Siebenhandl S, weinhäusel A. Systemic autoinflammatory diseases. J Autoimmun. 2020;109:102421. DOI: 10.1016/j.jaut.2020.102421.
3. Барабанова О.В., Коноплeва Е.А., Продеус А.П., щербина А.Ю. Периодические синдромы. Трудный пациент. 2007;5(2): 21-24. eLIBRARY ID: 16861557
4. Салугина С.О., Каменец Е.А., Федоров Е.С., Захарова Е.Ю., Каледа М.И. Результаты молекулярно-генетического скрининга мутаций генов NLRP3, TNFRSF1A, MVK у пациентов с аутовоспалительными заболеваниями и системным ювенильным артритом. Современная ревматология. 2017;11(3):33-43. DOI: 10.14412/1996-7012-2017-3-33-43
5. Barron K., Athreya B., Kastner D. Periodic fever syndromes and other inherited autoinflammatory diseases in: Cassidy jT, editor. Textbook of pediatric rheumatology. 7th ed. Elsevier Saunders; 2015.
6. Гатторно М. Аутовоспалительные заболевания у детей. Вопросы современной педиатрии. 2014;13(2):55-64. eLIBRARY ID: 21467355
7. Di Donato G, d'Angelo DM, Breda L, Chiarelli F. Monogenic Autoinflammatory Diseases: State of the Art and Future Perspectives. Int J Mol Sci. 2021;22(12):6360. DOI: 10.3390/ijms22126360.
8. Дубко М.Ф., Раупов Р.К., Канева М.А., Суспицын Е.Н., Костик М.М. Синдром дефицита мевалонаткиназы у ребенка раннего возраста: ключевые аспекты диагностики и лечения. Педиатрия. Журнал им. Г.Н. Сперанского. 2021;100(2):276-283. DOI: 10.24110/0031-403x-2021-100-2-276-283
9. Румянцев А.Г., Демина О.М. Аутовоспалительные заболевания: современная концепция патогенеза, клиники и диагностики. Педиатрия. Журнал им. Г.Н. Сперанского. 2020;99(3):211-219. DOI: 10.24110/0031-403x-2020-99-3-211-219
10. Favier LA, Schulert GS. Mevalonate kinase deficiency: current perspectives. Appl Clin Genet. 2016;9:101-10. DOI: 10.2147/TACG.S93933.
11. Munoz MA, jurczyluk j, Simon A, Hissaria P, Arts Rjw, et al. Defective Protein Prenylation in a Spectrum of Patients with Mevalonate Kinase Deficiency. Front Immunol. 2019;10:1900. DOI: 10.3389/fimmu.2019.01900.
12. Rodrigues F, Philit jB, Giurgea I, Anglicheau D, Roux jj, et al. AA amyloidosis revealing mevalonate kinase deficiency: A report of 20 cases including two new French cases and a comprehensive review of literature. Semin Arthritis Rheum. 2020;50(6):1370- 1373. DOI: 10.1016/j.semarthrit.2020.03.005.
13. Ozen S, Kuemmerle-DeschnerjB, Cimaz R, Livneh A, Quartier P, et al. International Retrospective Chart Review of Treatment Patterns in Severe Familial Mediterranean Fever, Tumor Necrosis Factor Receptor-Associated Periodic Syndrome, and Mevalonate Kinase Deficiency/Hyperimmunoglobulinemia D Syndrome. Arthritis Care Res (Hoboken). 2017;69(4):578- 586. DOI: 10.1002/acr.23120.
14. Козлова А.Л., Варламова Т.В., Зимин С.Б., Новичкова Г.А., щербина А.Ю. Опыт ведения больных с гипep-IGD-синдромом (синдромом дефицита мевалонаткиназы). Вопросы гематологии/онкологии и иммунопатологии в педиатрии. 2016;15(1):46–53. DOI: 10.20953/1726-1708-2016-1-46-53
15. Патрушева Ю.С., Бакрадзе М.Д. Синдром периодической лихорадки с дефицитом мевалонаткиназы (синдром гипериммуноглобулинемии D) у детей. Вопросы современной педиатрии. 2012;11(2):140-145. eLIBRARY ID: 17705237
16. Gattorno M, Sormani MP, D'Osualdo A, Pelagatti MA, Caroli F, et al.. A diagnostic score for molecular analysis of hereditary autoinflammatory syndromes with periodic fever in children. Arthritis Rheum. 2008;58(6):1823-32. DOI: 10.1002/art.23474.
Сомова Татьяна Михайловна, к.м.н., старший преподаватель кафедры детских болезней, Медицинский институт
Сургут
Сомова Т.М. Аутовоспалительное заболевание синдром гипериммуноглобулинемии D. Медицинский вестник Юга России. 2022;13(2):141-145. https://doi.org/10.21886/2219-8075-2022-13-2-141-145
Somova T.M. Autoinflammatory disease syndrome of hyperimmunoglobulinemia D. Medical Herald of the South of Russia. 2022;13(2):141-145. (In Russ.) https://doi.org/10.21886/2219-8075-2022-13-2-141-145
344022, г. Ростов-на-Дону, пер. Нахичеванский, 29
Ростовский государственный медицинский университет
Тел.: +7 918 571 0558
E-mail: journal@medicalherald.ru


































