



Contents
Scroll to:
https://doi.org/10.21886/2219-8075-2022-13-2-141-145
Scroll to:
Objective: present a clinical case of a rare autoinflammatory disease.
Materials and methods: an analysis of a clinical case of mevalonate kinase deficiency syndrome in an 8-year-old girl was carried out.
Results: mevalonate kinase deficiency syndrome (SDMC) is a rare autoinflammatory disease with an autosomal recessive inheritance mechanism. The severity of the disease correlates with the residual activity of the enzyme mevalonate kinase, which is involved in the biosynthesis of cholesterol and isoprenoids, which is necessary for the conversion of mevalonate into the final product. As a result, the body accumulates mevalonic acid, especially high levels of it are found in the urine. A relatively mild SDMC phenotype is distinguished: hyperimmunoglobulinemia syndrome D (first described as HIDS in 1984) and a severe variant – mevalonic aciduria. A little more than 300 patients with SDMC have been described in the world. The disease is manifested by intermittent fever, arthralgia, pharyngitis, cervical lymphadenitis, urticarial rash resembling urticaria, less often vasculitis-like manifestations in the form of petechial-purple elements. Diagnosis is based on the activity of the mevalonate kinase enzyme in the blood or skin cells, biochemical analysis of urine (high numbers of mevalonic acid), as well as genetic confirmation of a mutation in the mevalonate kinase gene. The main principles of therapy for autoinflammatory diseases are the control of clinical symptoms and inflammation in general, as well as the prevention of amyloidosis.
Conclusion: if an autoinflammatory process in the body is suspected, it is necessary to conduct a genetic study for timely diagnosis and the appointment of pathogenetic therapy to improve the quality of life and prevent complications.
Somova T.M. Autoinflammatory disease syndrome of hyperimmunoglobulinemia D. Medical Herald of the South of Russia. 2022;13(2):141-145. (In Russ.) https://doi.org/10.21886/2219-8075-2022-13-2-141-145
Various autoinflammatory diseases (AIDs) are currently being intensively studied. Molecular genetic testing of patients is of great importance for the diagnosis of such diseases, since the basis for their development is pathological mutations that cause disorders in the system of innate (antigen-nonspecific) immunity and the development of inflammation. An expert group consisting of 12 German and Austrian academic specialists has developed a diagnostic algorithm and principles for the treatment of auto-inflammatory processes [1].
Autoinflammation is considered part of the etiopathogenesis of such common diseases as Crohn's disease, erythema nodosum, sarcoidosis, ankylosing spondylitis, vasculitis and vasculitis syndromes, macrophage activation syndrome, monogenic vasculitis (STING-associated vasculitis and type II adenosine-deaminase deficiency (DADA2)), adult Still's disease, systemic lupus erythematosus, systemic juvenile idiopathic arthritis, gouty arthritis, and psoriasis (mutation of the CARD14 gene) [2].
Main AID characteristics:
Nowadays, more than 30 genes are known, mutations in which lead to the development of AIDs; the most common and well-studied are the NLRP3, TNFRSF1A, and MVK genes.
These genes cause the development of the following major monogenic AIDs: cryopyrin-associated periodic syndromes (CAPS); TNF-receptor-associated periodic syndrome (TRAPS); hyper-immunoglobulinemia D-syndrome (HIDS) [3-7].
Hereditary periodic fever (a typical violation of the function of inflammasomes):
Pyogenic diseases of the skin and bones (possible involvement of inflammasomes and other mechanisms):
Granulomatous diseases (violation of NF kB activation and signal transmission by IL-10):
Idiopathic syndromes + other nosological units (unclear pathogenesis, possible involvement of inflammasomes):
A decrease in the activity of mevalonate kinase leads to a deficiency of geranyl-geranyl-pyrophosphate (GGPP), one of the end products of mevalonic acid involved in the prenylation of intracellular G-proteins, and, as a consequence, leads to excessive activity of procaspase, synthesis of the precursor of interleukin-1 (IL-1)-beta and its active form [5-13].
A milder variant of mevalonate kinase deficiency, also known as hyper-IgD syndrome, is more common and is characterized by recurring episodes of unexplained fever without concomitant infection. Such attacks are accompanied by fatigue, chills, abdominal pain, swelling of the affected lymph nodes (lymphadenopathy), rash, joint inflammation (arthritis), and pain (arthralgia). Additional symptoms include nausea, diarrhea, vomiting, headaches, small mouth ulcers, and abnormal enlargement of the liver and spleen (hepatosplenomegaly). The rash is presented in the form of erythematous spots and papules. Some people have cough and inflammation of the posterior pharyngeal wall (pharyngitis). As a result of the long course of the disease, complications such as kidney amyloidosis, joint contractures, and abdominal adhesions develop [14-16].
The purpose of this study is to present a clinical observation of autoinflammatory disease in a patient.
Therefore, a clinical case of hyperimmunoglobulinemia D (mevalonate kinase deficiency syndrome) in an 8-year-old girl was analyzed.
Girl R., 8 years old (date of birth – November 24, 2013), born from the 4th pregnancy against the background of a burdened obstetric history (her mother had 2 miscarriages at an early stage), 2 urgent deliveries. Birth weight — 3660 g, Apgar score — 8/9 points. Preventive vaccinations according to an individual calendar (BCG, V1 against polio, V1–V2 against viral hepatitis B). The child suffered from the following diseases — chickenpox in 1 year 10 months, acute respiratory viral infections, follicular sore throat, acute bronchitis, acute tonsillitis, infectious mononucleosis. The heredity for hematological, immunological, and oncological diseases was not burdened.
The patient has been ill since the age of two, when, after chickenpox, a fever attack of up to 40 degrees first appeared, accompanied by an increase in the palatine tonsils, the appearance of purulent plaque on them, and an increase in cervical lymph nodes (Figure 1). This attack was regarded as an infectious process, stopped by antibacterial therapy and the use of nonsteroidal anti-inflammatory medicines (NSAIDs).
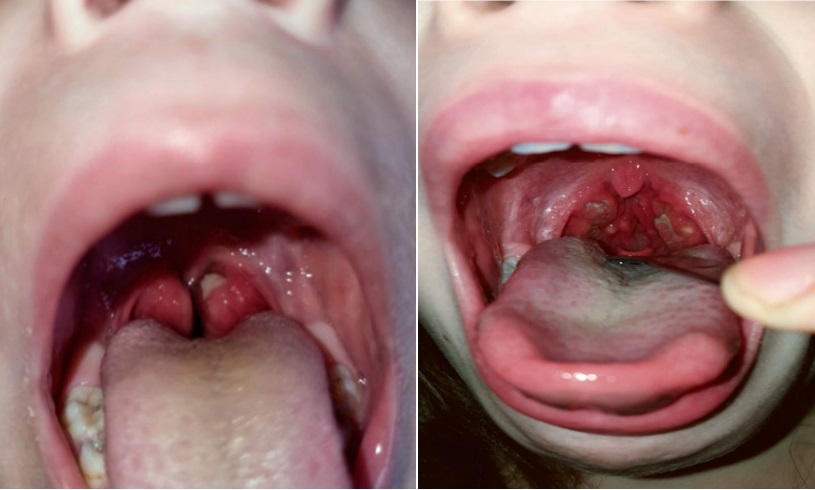
Рисунок 1. Налеты на небных миндалинах в приступный период (собственное наблюдение)
Figure 1. Raids on the palatine tonsils during the onset period (own observation)
However, then such episodes began to recur every 20–30 days with the duration of the attack 5–7 days. There was an intestinal syndrome in the form of abdominal pain, nausea, vomiting, loose stools, arthralgia, and an increase in inflammatory activity of the blood (ESR up to 28 mm/hour) began to join the temperature and increase in the tonsils.
Outside of the attack, the patient showed a satisfactory condition. Her physical development was harmonious according to the mesosomatic type. All the internal organs were without features. Physiological recovery was normal.
During the year, there was a positive effect during attacks in the form of fever relief after taking NSAIDs.
From the age of three, the patient has been observed by an immunologist with a diagnosis of Marshall syndrome; dysgammaglobulinemia was detected as well. During the attacks, she took glucocorticosteroids (GCS) (Prednisone orally 1 mg/kg with a positive effect). At the age of 6, she was consulted by a rheumatologist, therefore, immunological studies were carried out (extractable nuclear antibodies, antibodies to cardiolipin (total), antibodies to single-, double-stranded DNA, antibodies to cyclic citrullinated peptides, antibodies to phospholipids); all the indicators were within normal limits. There was not any data on autoimmune pathology. During the attacks, it was recommended to take GCS (metipred) (1.5 mg/kg). Further, due to a decrease in the response to GCS, the dose of Prednisone was gradually escalated to 2 mg/kg during attacks.
Taking into account the long course of the disease and the short intervals between attacks and the response only to an increase in the dose of GCS to 2 mg/kg at 7.5 years of age, a molecular genetic study was conducted, as a result of which a mutation in the MVK gene was detected in the 11th exon (V377I) and the 9th exon (I268T) in a heterozygous state. This confirms the diagnosis of mevalonate kinase deficiency syndrome.
In order to determine further management tactics, the patient was sent to the Department of Immunology of the Federal State Budgetary Institution “National Medical Research Center of Pediatric Hematology, Oncology and Immunology named after Dmitry Rogachev” of the Ministry of Health of Russia, where she was hospitalized in July 2021.
Upon admission, the somatic status was stable, there was moderate hypertrophy of the tonsils (1–2 degrees) with a single plaque.
Laboratory test results:
Results of instrumental research methods:
Medical conclusion: the examined biopsy material showed signs of active erosive ileitis, inactive reactive colitis. There were no signs of amyloidosis.
Consultation by an ophthalmologist — OU without pathological changes.
The diagnosis was “Primary immunodeficiency condition: autoinflammatory syndrome — mevalonate kinase deficiency syndrome (mutation in the MVK gene in exon 11 (V377I) and exon 9 (I268T) in a heterozygous state)”.
This disease is chronic in its nature; in the absence of control of inflammatory attacks, the risk of developing kidney amyloidosis and multiple organ failure and other life-incompatible complications is high.
Taking into account the severity of the disease course, as well as the presence of recurrent episodes of high fever in the patient, accompanied by an increase in the palatine tonsils, intestinal syndrome, nausea and arthralgia, as part of achieving and maintaining remission, the girl was prescribed a monoclonal inhibitor IL-1b Kanakinumab (Ilaris) at a dose of 75 mg once every 8 weeks, subcutaneously, according to vital indications.
Already after the first administration of the medicine, the attack-free interval increased to 50 days.
For all the children with periodic fever syndrome, a genetic study is indicated for the timely diagnosis of rare auto-inflammatory diseases and the appointment of pathogenetic therapy to improve the quality of life and prevent complications.
1. Hansmann S, Lainka E, Horneff G, Holzinger D, Rieber N, et al. Consensus protocols for the diagnosis and management of the hereditary autoinflammatory syndromes CAPS, TRAPS and MKD/HIDS: a German PRO-KIND initiative. Pediatr Rheumatol Online J. 2020;18(1):17. DOI: 10.1186/s12969-020-0409-3.
2. Krainer j, Siebenhandl S, weinhäusel A. Systemic autoinflammatory diseases. J Autoimmun. 2020;109:102421. DOI: 10.1016/j.jaut.2020.102421.
3. Barabanova O.V., Konopleva Ye.A., Prodeus A.P., Shcherbina A.YU. Periodicheskiye sindromy. Trudnyy patsiyent. 2007;5(2):21- 24. (In Russ.). eLIBRARY ID: 16861557
4. Salugina S.O., Kamenets E.A., Fedorov E.S., Zakharova E.Yu., Kaleda M.I. Results of molecular genetic screening of mutations in the NLRP3, TNFRSF1A, and MVK genes in patients with autoinflammatory diseases and systemic juvenile arthritis. Modern Rheumatology Journal. 2017;11(3):33-43. (In Russ.) DOI: 10.14412/1996-7012-2017-3-33-43
5. Barron K., Athreya B., Kastner D. Periodic fever syndromes and other inherited autoinflammatory diseases in: Cassidy jT, editor. Textbook of pediatric rheumatology. 7th ed. Elsevier Saunders; 2015.
6. Gattorno M. Autoinflammatory diseases in children. Current pediatrics (Moscow). 2014;13(2):55-64. (In Russ.). eLIBRARY ID: 21467355
7. Di Donato G, d'Angelo DM, Breda L, Chiarelli F. Monogenic Autoinflammatory Diseases: State of the Art and Future Perspectives. Int J Mol Sci. 2021;22(12):6360. DOI: 10.3390/ijms22126360.
8. Dubko M.F., Raupov R.K., Kaneva M.A., Suspitsyn E.N., Kostik M.M. Mevalonate kinase deficiency in an infant: key aspects of diagnosis and treatment. Pediatrics. Journal named after G.N.Speransky. 2021;100(2):276-283. (In Russ.). DOI: 10.24110/0031-403x-2021-100-2-276-283
9. Rumyantsev A.G., Demina O.M. Autoinflammatory diseases: a modern concept of pathogenesis, clinic and diagnosis. Pediatrics. Journal named after G.N.Speransky. 2020;99(3):211-219. (In Russ.). DOI: 10.24110/0031-403x-2020-99-3-211-219
10. Favier LA, Schulert GS. Mevalonate kinase deficiency: current perspectives. Appl Clin Genet. 2016;9:101-10. DOI: 10.2147/TACG.S93933.
11. Munoz MA, jurczyluk j, Simon A, Hissaria P, Arts Rjw, et al. Defective Protein Prenylation in a Spectrum of Patients with Mevalonate Kinase Deficiency. Front Immunol. 2019;10:1900. DOI: 10.3389/fimmu.2019.01900.
12. Rodrigues F, Philit jB, Giurgea I, Anglicheau D, Roux jj, et al. AA amyloidosis revealing mevalonate kinase deficiency: A report of 20 cases including two new French cases and a comprehensive review of literature. Semin Arthritis Rheum. 2020;50(6):1370- 1373. DOI: 10.1016/j.semarthrit.2020.03.005.
13. Ozen S, Kuemmerle-Deschner jB, Cimaz R, Livneh A, Quartier P, et al. International Retrospective Chart Review of Treatment Patterns in Severe Familial Mediterranean Fever, Tumor Necrosis Factor Receptor-Associated Periodic Syndrome, and Mevalonate Kinase Deficiency/Hyperimmunoglobulinemia D Syndrome. Arthritis Care Res (Hoboken). 2017;69(4):578-586. DOI: 10.1002/acr.23120.
14. Kozlova A.L., Varlamova T.V., Zimin S.B., Novichkova G.A., Shcherbina A.Yu. Experience gained in the treatment of patients with hyper-IgD syndrome (mevalonate kinase deficiency). Vopr. gematol./onkol. immunopatol. pediatr. (Pediatric Haematology/ Oncology and Immunopathology). 2016;15(1):46–53. (in Russ.). DOI: 10.20953/1726-1708-2016-1-46-53
15. Patrusheva j.S., Bakradzeе M.D. Periodic fever syndrome (mevalonate kinase deficiency, hyperimmunoglobulinemia D syndrome) in children. Current pediatrics (Moscow). 2012;11(2):140-145. (in Russ.). eLIBRARY ID: 17705237
16. Gattorno M, Sormani MP, D'Osualdo A, Pelagatti MA, Caroli F, et al.. A diagnostic score for molecular analysis of hereditary autoinflammatory syndromes with periodic fever in children. Arthritis Rheum. 2008;58(6):1823-32. DOI: 10.1002/art.23474.
Tatyana M. Somova, Cand. Sci. (Med.), Senior Lecturer, Children’s Diseases Department, Medical Institute
Surgut
Somova T.M. Autoinflammatory disease syndrome of hyperimmunoglobulinemia D. Medical Herald of the South of Russia. 2022;13(2):141-145. (In Russ.) https://doi.org/10.21886/2219-8075-2022-13-2-141-145
29, Nakhichevansky Lane, Rostov-on-Don, 344002
Rostov State Medical University
Тel.: +79185710558
e-mail: journal@medicalherald.ru


































