



Содержание
Перейти к:
https://doi.org/10.21886/2219-8075-2021-12-2-28-35
Перейти к:
Цель: изучить основные аспекты оказания помощи детям с орфанными заболеваниями в Российской Федерации. Материалы и методы: для реализации указанной цели были изучены наиболее актуальные литературные источники, содержание которых освещало представления об орфанных заболеваниях в различных странах мира и в РФ, а также тактику и регулирование механизмов помощи пациентам с редкими болезнями. Результаты: исследование показало, что далеко не во всех странах существует законодательное регулирование оказания помощи орфанным пациентам и наиболее передовыми в этом отношении являются США и страны Западной Европы, где определены четкие критерии определения орфанной патологии, а также предпринимается ряд мер для повышения качества медицинского обслуживания больных с редкими заболеваниями. Эти мероприятия направлены не только на улучшение работы в системе здравоохранения, но и поощряют фармацевтические компании к разработке и производству лекарственных средств, а также способствуют научным исследованиям в указанной области. рассмотренный в статье клинический случай дает представление о редких заболеваниях, сложностях их диагностики, тяжести течения и лекарственных средствах, которые необходимы для помощи пациентам. Выводы: орфанные заболевания в последние десятилетия стали объектом пристального внимания со стороны системы здравоохранения и национального законодательства. Их чрезвычайно низкая распространённость в человеческой популяции создаёт затруднения с своевременной постановкой диагноза, оказанием квалифицированной медицинской помощи и лекарственным обеспечением.
Шашель В.А., Фирсова В.Н., Трубилина М.М., Подпорина Л.А., Фирсов Н.А. Орфанные заболевания и связанные с ними проблемы. Медицинский вестник Юга России. 2021;12(2):28-35. https://doi.org/10.21886/2219-8075-2021-12-2-28-35
Shashel V.A., Firsova V.N., Trubilina M.M., Podporina L.A., Firsov N.A. Orphan diseases and associated problems. Medical Herald of the South of Russia. 2021;12(2):28-35. (In Russ.) https://doi.org/10.21886/2219-8075-2021-12-2-28-35
К орфанной патологии относят заболевания, очень редко диагностируемые в человеческой популяции. Они могут иметь прогрессирующее течение и нести прямую угрозу для жизни пациента, если он не получает надлежащего лечения. Часто такие болезни заканчиваются инвалидностью. Критерием определения указанной патологии служит её распространение, которое, согласно требованиям Евросоюза, не должно превышать 1 случай на 2 тыс. человек. В США это соотношение составляет 1:1250.
В РФ к орфанным болезням относят патологию, обнаруживаемую с частотой 1:10000 человек и реже. Существует несколько определений орфанных заболеваний в русскоязычной литературе. Наиболее часто используемое характеризует их, как редкие заболевания, обнаруживаемые с невысокой частотой, угрожающие жизни или неуклонно прогрессирующие патологии, которые при отсутствии лечения могут вызвать летальный исход или привести к инвалидности.
По определению ФЗ № 323 от 21.11.2011 г. «Об охране здоровья граждан», к орфанной относят такую патологию, которая обнаруживается не чаще, чем у 10 из 100 тыс. человек.
Распространённость орфанных заболеваний в разных странах мира заметно варьируется. Например, у полинезийцев, живущих на Гавайях, распространённость муковисцидоза составляет 1:90000 детей, а в России эта пропорция равняется 1:10000. Иногда такая ситуация может быть основой для удаления какой-либо нозологии из национального перечня, если её частота превосходит пороговый уровень, приводя к количественному несоответствию статистических данных по определённым пациентам в разных странах1.
Из-за большого перечня орфанных заболеваний (по данным ВОЗ, >7000 наименований), общее число таких больных превышает 5% среди всего населения планеты2 3.
Распространённость заболеваний указанной группы (согласно документам Европейского общества) представлена в табл. 1 [1].
Таблица / Table1
Частота диагностики отдельных нозологических форм (по данным Eurordis, 2012, с дополнениями)
Indicators of prevalence of separate groups of hereditary diseases (according to Eurordis, 2012 with additions)
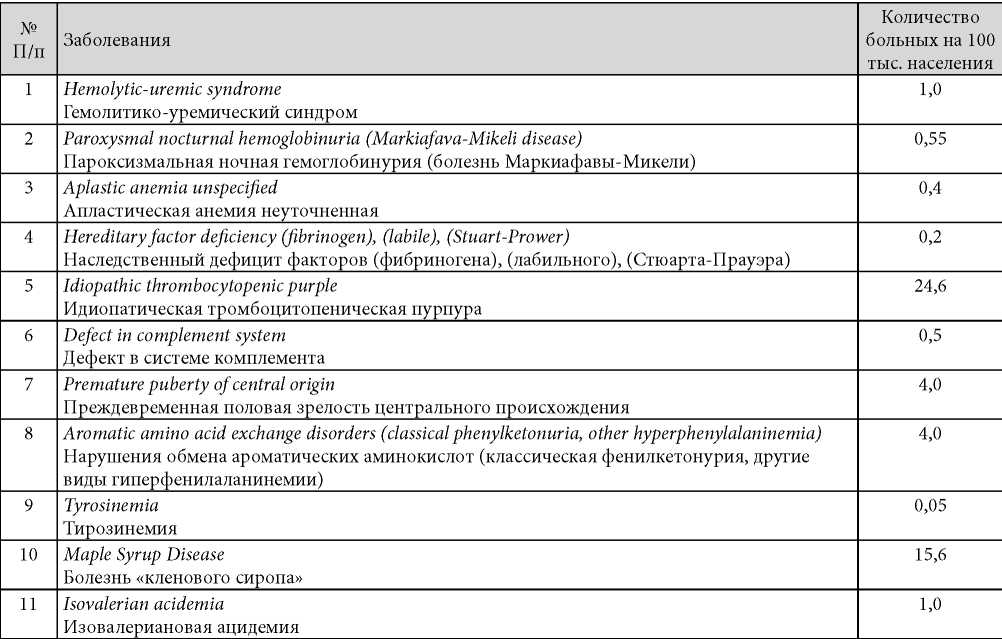

В России выделено 216 групп болезней, в которые входят от одного до восьми кодов заболеваний по МКБ10. Около 1000 нозологических форм включено в этот перечень. Список опубликован на сайте Минздрава РФ (www.rosminzdrav.ru/documents/8048-perechen-redkihorfannyhzabolevaniy).
Федеральный закон № 323 от 21.11.2011 г. (ст. 44) устанавливает несколько моментов, касающихся редких заболеваний. Минздрав формирует перечень редких (орфанных) заболеваний на основании статистических данных и размещает его на своём сайте. Некоторые заболевания из указанного списка собраны в Перечне нозологий, сопровождающихся угрозой для жизни, а также хронических орфанных патологий с прогрессирующим течением, способных привести к инвалидизации, и перспективой укорочения времени жизни пациентов. Перечень установлен Правительством РФ от 26.04.2012 г. № 403 (табл. 2) [1].
Таблица / Table 2
Список орфанных и наследственных болезней, согласно Постановлению Правительства РФ № 403 от 26.04.2012 г.
The list the zhizneugrozhayushchikh and chronically progressing rare (orphan) and hereditary diseases according to Resolution of the Government of the Russian Federation No. 403 of 26.04.2012
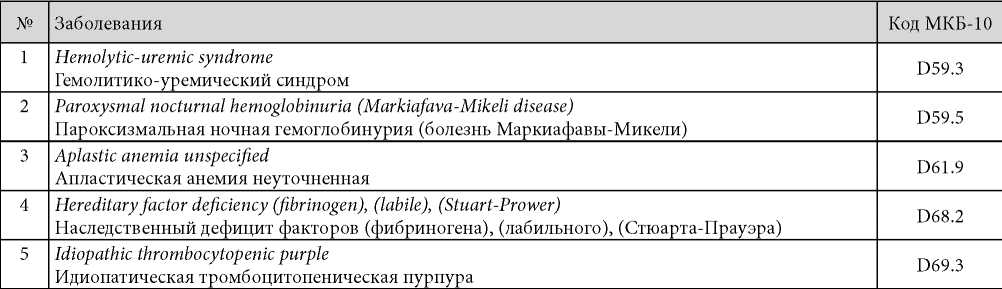

Было выяснено, что в отдельных странах имеются собственные определения термина «орфанные заболевания» и свой перечень патологии, которая относится к указанной группе. Количество нозологических единиц из списка орфанных заболеваний варьируется от 214 (в России) до 6000 – 8000 (в странах Запада).
В России с 1985 г. в родильных домах начал проводиться скрининиг новорождённых в целях диагностики генетически обусловленной ферментопатии (фенилкетонурии), которая в отсутствии лечения приводит к тяжёлому необратимому заболеванию нервной системы с развитием умственной отсталости. В 1993 г. началась реализация программы «Дети России», которая предполагает обследование новорождённых на наличие у них признаков снижения функции щитовидной железы. Проект «Здоровье», регламентированный приказом Минздравсоцразвития России от 22.03.2006 г. № 185 «О массовом обследовании новорождённых детей на наследственные заболевания», позволил начать комплексную диагностику генетической патологии новорожденных. В частности, проект охватывает тестирование таких болезней, как муковисцидоз, фенилкетонурия, адреногенитальный синдром и др. Также детям проводят исследование слуха и при его снижении ребенка направляют на расширенную диагностику и лечение тугоухости.
План неонатального тестирования в обязательном порядке включает несколько стадий: 1) отбор биоматериала для диагностики у всех новорождённых и транспортировку его в лабораторию; 2) быстрое предварительное тестирование; 3) уточняющее исследование того материала, который дал положительный результат на предварительном тестировании; 4) лечение пациентов и регулярный мониторинг качества лечения; 5) семейная медико-генетическая консультация.
Всеобщее скрининговое тестирование новорождённых вместе с пренатальным исследованием и семейной медико-генетической консультацией служит мощным средством профилактики развития генетических заболеваний у новорождённых и их распространения в человеческой популяции.
Орфанные заболевания из-за своей низкой распространенности создают много сложностей для пациентов как на этапе диагностики, так и на этапе лечения. Клинический опыт врача ограничен в отношении редких болезней, и на установление истинной причины недуга может уйти достаточно много времени.
Во всех странах очень мало действующих медицинских центров, специализирующихся на лечении редко встречающейся патологии, а фармацевтические компании крайне неохотно берутся за разработку новых орфанных препаратов, поскольку рынок сбыта для них невелик.
Всё перечисленное ущемляет права таких пациентов на получение качественной медицинской помощи и системы здравоохранения разных государств стремятся выработать наиболее эффективный подход в отношении орфанных больных.
Определения орфанных заболеваний в разных странах заметно различаются. В США принято считать, что в эту категорию входит патология, поражающая не более 200000 человек на данный момент времени. В подавляющем большинстве других стран в основе определения лежит относительное распространение той или иной нозологии в человеческой популяции [2].
На сайте Orphanet представлена номенклатура редких заболеваний, которая применяется в информационных системах здравоохранения и в исследованиях: каждой нозологической единице присваивается свой уникальный и постоянный идентификатор — номер ORPHA. В Orphanet используется европейское определение редкого заболевания, как оно определено в Регламенте Европейского союза о лекарственных средствах для лечения редких заболеваний (1999 г.). Согласно правилам, принятым в ЕС, орфанное заболевание поражает не более 1 человека на 2000 в европейской популяции.
Номенклатура редких заболеваний Orphanet состоит из списка заболеваний с уменьшающейся распространённостью, основные характеристики включают группы нарушений и расстройств, их подтипы [3].
Несмотря на то, что речь идёт об очень редких болезнях, общий перечень орфанных заболеваний столь велик (по разным оценкам, 6000 – 8000), что во всём мире ими страдает примерно 400 млн человек. 80% таких заболеваний имеют генетическую природу, по степени тяжести они варьируются от лёгких до угрожающих жизни. Большое число указанных заболеваний требует раннего начала лечения, дорогостоящего медицинского ухода и больших материальных издержек. Например, у взрослых с расщелиной позвоночника (spina bifida) затраты на медицинские услуги превышают в 3 – 6 раз таковые для здоровых людей. По данным 2014 г., в США средняя стоимость медицинских препаратов для людей с орфанными заболеваниями составили 137 782 доллара на одного пациента [2].
Большую проблему в лечении указанных больных представляет географический разброс таких людей по большой территории, что затрудняет их концентрацию в одном специализированном медицинском учреждении, где они могли бы получить качественную помощь.
Другим фактором, неблагоприятствующим больным с орфанными заболеваниями, является то, что фармацевтическим компаниям невыгодно разрабатывать и производить лекарства, потребность в которых заведомо очень мала. В 1983 г. Конгресс США принял Закон об орфанных лекарственных средствах. Этот закон поощрял производство подобных лекарств, предоставляя финансовые стимулы для фармацевтической промышленности, чтобы компенсировать потенциальные потери сбыта лекарств на таком маленьком рынке [4]. Кроме того, в 2001 г. были приняты «Поправки 2001 г. об оказании помощи сообществу в области мышечной дистрофии» [2]. Цель данной поправки состояла в усовершенствовании методов скрининга, улучшении связей между специализированными лечебными учреждениями и разработке образовательных программ, ориентирующих специалистов в особенностях различных видов мышечной дистрофии.
Большую роль в борьбе с редкими болезнями играют профилактические мероприятия, организованные на общенациональном уровне. В 1998 г. вступило в силу постановление FDA (Food and Drug Administration), которое предписывало добавлять фолиевую кислоту в продукты из злаковых зерен. Это способствовало снижению дефицита фолиевой кислоты в организме беременных женщин и предотвращало появление врожденных дефектов развития нервной трубки у новорожденных [5].
В настоящее время в 86 странах мира продукты с зерновыми злаками обогащаются фолиатами [2]. Европейский союз демонстрирует комплексный подход в борьбе с редкими заболеваниями, который распространяется на все страны ЕС. Так, во всех 28 странах практикуется общее определение орфанных заболеваний, а деятельность, с ними связанная, определяется «Регламентом лекарственных средств для орфанных заболеваний № 141». Данное постановление способствует научным исследованиям по орфанной патологии и разработке новых орфанных лекарственных средств. Также в ЕС разработан единый национальный план по редким заболеваниям (EUROPLAN) для облегчения создания местных национальных стратегий помощи больным с орфанной патологией. Эти планы имеют собственный бюджет и четкие сроки реализации.
В странах Европы предоставляются налоговые льготы фармацевтическим компаниям, разрабатывающим орфанные препараты, к тому же они пользуются так называемой эксклюзивностью рынка в течении 10 лет после начала выхода нового медикамента в продажу.
Таким образом, несмотря на маленький рынок сбыта, компания оправдывает свои издержки на разработку нового лекарства [2]. В 2009 г. Европейский совет министерств здравоохранения принял план действий по редким заболеваниям, в котором положил начало разработке и принятию национальных планов по редким заболеваниям к концу 2013 г.
В 2011 г. программа действий по редким заболеваниям активно обсуждалась Директивой ЕС №м24, в ней говорилось о применении прав пациентов в трансграничном здравоохранении. Директива определила основные правила обращения пациентов за медицинской помощью в другие страны ЕС (кроме резидентной). Это способствовало развитию сотрудничества между системами здравоохранения стран ЕС, в том числе посредством внедрения Европейских справочных систем. Последние были созданы для того, чтобы поддержать объединение европейских экспертных центров и специалистов в разных странах для обмена знаниями и определения альтернативных вариантов лечения, содействия исследованиям и распространению инноваций в целях оказания наилучшей медицинской помощи пациентам с орфанными заболеваниями [6].
Законодательное регулирование в отношении орфанных заболеваний существует в РФ с ноября 2011 г. Согласно утвержденному закону «Об основах охраны здоровья граждан в Российской Федерации» от 2011 г. на плечи регионов РФ ложится задача по снабжению орфанных больных соответствующими лекарственными препаратами.
Постановление Правительства Российской Федерации № 403 предусматривает ведение Федерального регистра пациентов с орфанной патологией, приводящей к снижению продолжительности жизни и инвалидизации. Регистр был создан с целью обеспечения орфанных больных необходимыми лекарственными препаратами и компонентами лечебного питания, он содержит персональную информацию, включая паспортные данные, СНИЛС, поставленный диагноз и др.
В РФ в настоящее время реализуется программа «Семь нозологий». Её план подразумевает централизованную закупку Государством необходимых лекарственных препаратов для больных, страдающих в том числе и орфанными заболеваниями (болезнь Гоше, муковисцидоз, гипофизарный нанизм и гемофилия). При этом закупка лекарств для детей финансируется из региональных бюджетов.
Существует ряд затруднений, с которыми имеют дело как сами орфанные больные, так и медицинские специалисты, оказывающие им помощь. Можно перечислить наиболее актуальные из указанных проблем:
Несколько недавно принятых документов, в том числе Постановление Правительства РФ от 26.11.2018 № 1416 «Правила организации обеспечения лекарственными препаратами» и «Правила ведения федерального регистра» дают возможность расширить базу данных по орфанным пациентам, сделать более доступным получение лекарственных препаратов, помогающих поддерживать функционирование организма на приемлемом уровне. Кроме того, эти нововведения предусматривают дополнительное обучение врачей и других медицинских специалистов навыкам и приёмам ведения пациентов с орфанной патологией: для этого разрабатываются образовательные программы и специализированные курсы повышения квалификации, врачи могут участвовать в обсуждении различных аспектов существующего законодательства, имеющего отношение к данной проблеме. Документы также нацелены на привлечение инвесторов для разработки и внедрения отечественных орфанных препаратов.
Российские граждане с орфанными заболеваниями очень часто не имеют возможности реализовать своё право на лекарственное обеспечение, поскольку медикамент еще не разработан, либо не зарегистрирован в РФ. Такие лекарства, как правило, очень дороги, а государство не может обеспечить больному полную оплату. Норма финансирования лекарственного снабжения пациентов, страдающих орфанной патологией, за счёт государственных средств очень часто делает лечение пациентов крайне затруднительным.
Ниже приведен клинический случай ребенка с орфанным заболеванием (наследственными болезнями обмена), проживающего на территории Краснодарского края.
Ребенок Г., 2014 г.р., находившийся на обследовании и лечении в ГБУЗ ДККБ. Из анамнеза: ребенок от первой беременности, проходившей в условиях токсикоза I степени в I триместре. Роды в срок, физиологические. Обратился на приём к участковому педиатру с длительным течением респираторной инфекции, ребенку было проведено биохимическое исследование крови, выявлено значительное повышение уровней аланинаминотрансферазы (АлАТ) до 145 Ед/л, аспартатаминотрансферазы (АсАТ) до 586 Ед/л. Исключены вирусные гепатиты. Ребенок был госпитализирован в стационар с диагнозом «Гепатит неуточненной этиологии (неинфекционный) с целью дальнейшего лечения и установления диагноза». Получал терапию гепатопротекторами, при этом отмечались жалобы на мышечную слабость, трудности при подъёме по лестнице, редкие головные боли. Объективно: состояние средней тяжести по основному заболеванию, самочувствие удовлетворительное. Физическое развитие ниже медианного уровня, гармоничное: рост — 88 см (10 – 25 перцентиль), вес — 12 кг (25 – 50 перцентиль), окружность головы — 49 см (25 – 50 перцентиль). Кожные покровы чистые, бледные. Мышечный тонус с тенденцией к гипотонии. В лёгких дыхание везикулярное, хрипов нет. Сердечные тоны ясные, ритмичные, выслушивается систолический шум на верхушке и в V точке. Живот при пальпации мягкий, болезненность отсутствует. Нижняя граница печени выходит из-под границы реберной дуги на 3,5 см по правой срединно-ключичной линии. Селезёнка не пальпируется. Общемозговые и менингеальные симптомы отсутствуют. Лабораторно: биохимический анализ крови: АсАТ — 395МЕ/л, АлАТ — 123 МЕ/л, лактатдегидрогеназа (ЛДГ) — 1357 Ед/л, креатининфосфокиназа (КФК) — 825Ед/л. Исследование иммуноглобулинов крови: иммуноглобулин Е — 409,7 МЕ/мл. В связи с подозрением на болезнь Помпе было проведено исследование крови методом тандемной масс-спектрометрии, при этом выявлено снижение активности альфа глюкозидазы (0,63 мкмоль/л/час при норме 1,0 — 25,0 мкмоль/л/час). При проведении молекулярно-генетического исследования методом прямого автоматического секвенирования проведен полный анализ гена GAA выявлено изменение нуклеотидной последовательности с.-32 – 13Т> G/c.-45T> G в гетерозиготном состоянии, описанное в международной базе данных по мутациям (CS941489), выявлено изменение нуклеотидной последовательности с.584Т> А, приводящей к замене р.Leu195Term в гетерозиготном состоянии. Ребенку был выставлен диагноз «Болезнь Помпе (гликогеноз II типа). Миопатический синдром». По жизненным показаниям, был назначен генно инженерный ферментозаменяющий препарат Майозайм (alglucosidasealfa, Genzyme Ireland Limited, Ирландия), который показан к длительному применению при всех формах болезни, — единственное патогенетическое средство для лечения пациентов с этим тяжёлым прогрессирующим наследственным заболеванием (1 раз в 2 недели).
Таким образом, прогноз болезни варьируется в зависимости от времени манифестации и выраженности симптомов. Для раннего лечения нужно своевременно диагностировать заболевание, а для диагностики болезни Помпе генетическое исследование не является ключевым методом, достаточно определение активности фермента GAA методом тандемной масс-спектрометрии.
Рассмотренный пример указывает на то, что орфанные заболевания часто представляют собой тяжёлые, угрожающие жизни состояния, и если их не лечить, то возможны летальный исход или тяжёлая инвалидизация. Это требует выработки специальной стратегии по оказанию помощи пациентам с редкими заболеваниями.
Ситуация с орфанными болезнями в России требует дальнейших разработки, модернизации и регулирования со стороны национального законодательства и системы здравоохранения. Необходимо улучшать взаимодействие между различными медицинскими учреждениями и вырабатывать общий план помощи и взаимодействия пациентам с редкими заболеваниями.
Огромное значение имеет улучшение доступности лекарственных препаратов для указанной группы больных. Необходимо перенимать опыт Евросоюза в отношении тактики взаимодействия с фармацевтическими компаниями, предоставляя им рыночные преференции при разработке орфанных препаратов.
Учитывая небольшой опыт врачей в отношении рассматриваемых заболеваний, возможно организовать больше лекционного материала по редкой патологии в составе курса повышения квалификации для медицинских специалистов.
В настоящее время существует необходимость интеграции планов и исследований по орфанным заболеваниям на международном уровне. Это могло бы изменить фрагментарный подход к решению проблемы и скоординировать общие усилия медицинского сообщества в отношении статистического учёта, поиска эффективного лечения, разработки скрининговых систем диагностики и информационной помощи врачам и пациентам.
1. Cайт Минздрава РФ: [Электронный ресурс]. – URL: http://www.rosminzdrav.ru/documents/8048-perechen-redkih-orfannyhzabolevaniy
2. Орфанные – значит, сиротские // [Электронный ресурс]. – Режим доступа: http://www.miloserdie.ru/articles/orfannye-znachit-sirotskie
3. Здравоохранение в России 2010: доклад.- Формулярный комитет РАМН. – М.: НЬЮДИАМЕД, 2011. – 165 с. [Электронный ресурс]. – Режим доступа: webmed.irkutsk.ru/doc/pdf/form2010.pdf
1. Подвязникова М.В. Правовое регулирование лекарственного обеспечения лиц, страдающих орфанными заболеваниями, в субъектах российской Федерации. // Российский ежегодник трудового права. – 2014. - № 9. – с. 606 - 615. eLIBRARY ID: 22480673
2. Ягудина Р.И., Королева И.И. Редкие заболевания и орфанные лекарственные средства. - М.: изд. «МИА»; 2015.
3. Aghajanzadeh M., Asgary M.R., Mesbah A., Hemmati H., Delshad M.S.E., et al. Giant thymolipoma of mediastinum and neck - initially misdiagnosed as liposarcoma by core needle biopsy. // J Family Med Prim Care. – 2018. – V.7(5). – P. 1079-1082. DOI: 10.4103/jfmpc.jfmpc_228_17
4. Cannizzo S., Lorenzoni V., Palla I., Pirri S., Trieste L., et al. Rare diseases under different levels of economic analysis: current activities, challenges and perspectives. // RMD Open. – 2018. – V.4(Suppl 1). – P. e000794. DOI: 10.1136/rmdopen-2018-000794
5. Cheung R.Y., Cohen J.C., Illingworth P. Orphan drug policies: implications for the United States, Canada, and developing countries. // Health Law J. – 2004. – V. 12. – P. 183-200. PMID: 16539081.
6. Goralski J.L., Lercher D.M., Davis S.D., Dellon E.S. Eosinophilic esophagitis in cystic fibrosis: a case series and review of the literature. // J Cyst Fibros. – 2013. – V. ;12(1). – P. 9-14. DOI: 10.1016/j.jcf.2012.09.002
Шашель Виктория Алексеевна, д.м.н., проф., заведующая кафедрой педиатрии №1
Краснодар
Фирсова Виолетта Николаевна, к.м.н., доцент кафедры педиатрии №1
Краснодар
Трубилина Марина Михайловна, к.м.н., доцент кафедры педиатрии №1
Краснодар
Подпорина Людмила Анатольевна, ассистент кафедры педиатрии №1
Краснодар
Фирсов Никита Алексеевич, учащийся
Краснодар
Шашель В.А., Фирсова В.Н., Трубилина М.М., Подпорина Л.А., Фирсов Н.А. Орфанные заболевания и связанные с ними проблемы. Медицинский вестник Юга России. 2021;12(2):28-35. https://doi.org/10.21886/2219-8075-2021-12-2-28-35
Shashel V.A., Firsova V.N., Trubilina M.M., Podporina L.A., Firsov N.A. Orphan diseases and associated problems. Medical Herald of the South of Russia. 2021;12(2):28-35. (In Russ.) https://doi.org/10.21886/2219-8075-2021-12-2-28-35
344022, г. Ростов-на-Дону, пер. Нахичеванский, 29
Ростовский государственный медицинский университет
Тел.: +7 918 571 0558
E-mail: journal@medicalherald.ru


































