



Содержание
Перейти к:
https://doi.org/10.21886/2219-8075-2020-11-4-6-23
Перейти к:
На сегодняшний день жировая ткань перестала восприниматься только как энергетическая субстанция с присущими ей свойствами в виде терморегуляции и механической защиты, известными с начала двадцатого века. Жировая ткань — это полноценный эндокринный орган, который распределен по всему организму. Полноценность его работы напрямую влияет на энергетический баланс не только посредством участия в обмене углеводов и жиров, но и продукцией множества адипокинов, общим числом более 600, известных на сегодняшний день. В данном обзоре разбирается причинно-следственная связь субклинического или системного воспаления жировой ткани с избытком энергетических ресурсов, инсулинорезистентностью, лептином, адипонектином, метаболитами эстрогенов и одним из наиболее провосполительных цитокинов интерлейкином 6. Также внимание уделено связи рака предстательной железы и ожирения как неоднозначной связи из-за максимально прикованного к тестостерону внимания. Дальнейшее изучение жировой ткани позволит установить конкретные патофизиологические механизмы ответственные за развитие не только нарушений углеводного обмена, но и ряда других систем ввиду не до конца понятного системного действия адипокинов и связанных с ними медиаторов воспаления у лиц с ожирением. Системный поиск литературы проводился по базам данных Medline, Scopus, Web of Science и elibrary.
Павлова З.Ш., Голодников И.И. Ожирение = воспаление. Патогенез. Чем это грозит мужчинам? Медицинский вестник Юга России. 2020;11(4):6-23. https://doi.org/10.21886/2219-8075-2020-11-4-6-23
Pavlova Z.Sh., Golodnikov I.I. Obesity = inflammation. Pathogenesis. How does this threaten men? Medical Herald of the South of Russia. 2020;11(4):6-23. (In Russ.) https://doi.org/10.21886/2219-8075-2020-11-4-6-23
«Внезапная смерть, более характерна для тучных, чем для худых»
Гиппократ, IV век до н.э.
В начале XXI в. на фоне результатов экспериментальных и клинических исследований, возникла гипотеза, а потом и понимание, что ожирение приводит к субклиническому системному воспалению, на фоне которого развиваются последующие коморбидные состояния: сердечно-сосудистые заболевания, сахарный диабет 2 типа, нарушение обмена мочевой кислоты, нарушение баланса половых гормонов, неалкогольная жировая болезнь печени, онкологические заболевания, с которыми сталкиваются врачи различных специальностей, на протяжении многих лет с той или иной степенью успешности, занимающиеся борьбой с многочисленными следствиями избыточно накопленной жировой ткани. И чем большее количество времени продолжается эта борьба, тем более выражены макро- и микрососудистые осложнения и тем менее привержен пациент к терапии.
Сейчас ученые уже активно обсуждают воспаление жировой ткани и эффективные меры борьбы с ним. Его называют по-разному, ввиду его субклинического течения, например, паравоспалением или низкоуровневым хроническим воспалением, метавоспалением и пр. Так ученые пытаются отобразить особенности воспаления жировой ткани. Стало понятно, что многие средства, которые использовались ранее и применяются сейчас при развитии атеросклероза, сердечно-сосудистых заболеваний или метаболического синдрома, оказывали свое действие через подавление выраженности воспаления. Но даже пять лет назад заявление о воспалении жировой ткани воспринималось во врачебной среде осторожно, если не сказать скептически.
Немного исторических фактов:
Воспаление жировой ткани характеризуется клеточной инфильтрацией, нарушением микроциркуляции, фиброзом, нарушением секреции адипокинов, абсолютным или относительным повышением в крови неспецифических маркеров воспаления, таких как С-реактивный белок, фибриноген, лейкоциты и прежде всего моноциты [3][4][5]. Факторы воспаления или цитокины активируют фосфолипазу А2, которая способствует синтезу из фосфолипидов арахидоновой кислоты, активной участницы воспалительного процесса, которая в свою очередь повышает активность ферментов циклооксигеназы и липооксигеназы, а те стимулируют синтез простагландинов серии 2, 4 и лейкотриенов1. Развиваются те пять стадий воспаления, которые подробно изучаются в курсе физиологии, отображенные на рис. 1.

Если посмотреть на рис. 2, то кроме каскада факторов воспаления в левой части рисунка видна и правая часть, отображающая еще одну важнейшую составляющую воспаления, — оксидативный стресс. И, как и должно быть при воспалении, цитокины стимулируют синтез активных форм кислорода, а активные формы кислорода, в свою очередь, стимулируют синтез цитокинов. Порочные круги — это визитная карточка системы воспаления.

Представление о жировой ткани, сугубо, как о хранилище энергетических ресурсов, утрачено навсегда. Жировая ткань приобрела статус эндокринного органа, способного продуцировать множество метаболически активных веществ, способных нарушить или поддерживать гомеостаз и их действие не ограничивается пределами синтезировавшей их ткани.
Что из себя представляет жировая ткань и из чего состоит? Развивается жировая ткань изначально из мезодермы (мезенхимальные стволовые клетки) и состоит из трех основных компонентов: из адипоцитов, клеток стромы и экстрацеллюлярного матрикса. В свою очередь строма представлена фибробластами, гладкомышечными, тучными и эндотелиальными клетками. Экстрацеллюлярный матрикс, в большей степени представлен фибриллярным коллагеном 1 и 3 типов, также гликопротеинами - ламинином, фибронектином и эластином [3][4][5][6].
В 95 % случаев всё начинается с того, что человек употребляет больше энергетических ресурсов, то есть он ест больше пищи, чем тратит калорий на физическую активность и метаболические процессы в организме. Съеденные лишние калории утилизируются в виде триглицеридов в адипоцитах, способствуя увеличению размера жировых клеток, то есть прогрессирует гипертрофия адипоцитов [7]. Рис. 3 очень четко отражает хронологию событий: когда человек ест без избытка калорий, его адипоциты нормального размера и нормально кровоснабжаются. Когда пища поступает в избытке, адипоциты увеличиваются до тех пор, «укладывая в себя жировые запасы», пока они могут нормально кровоснабжаться. Бесконечно расти клетки не могут, и после возникновения микрогипоксии наступает этап, который будет детально описан ниже. В последующем увеличивается и количество жировых клеток, то есть кроме гипертрофии клеток развивается процесс гиперплазии жировой ткани, то есть увеличивается и количество жировых клеток. Большинство пациентов на вопрос врача о количестве и качестве съедаемого отвечают, что их рацион достаточно скуден и порции существенно меньше, чем у каких-то друзей или знакомых, при этом эти друзья не имеют лишнего веса, потому что их метаболизм отличается особенностями, позволяющими «сжигать» избыток калорий. Чаще всего они искренне заблуждаются, хотя может показаться, что они лукавят. И конечно же, мы все имеем особенности, в том числе и в количестве усвояемых калорий. При этом закон сохранения массы Ломоносова – Лавуазье, не оставляет надежд на то, что, не употребляя лишних калорий, люди накапливают лишнюю жировую ткань.

Экспериментальные исследования на мышах, которых кормили высококалорийной пищей, стимулирующей воспаление, на разных этапах демонстрировали разный клеточный состав тканей. Например, миграция нейтрофилов и Т лимфоцитов в жировую ткань стартовала на 3 – 7 дней раньше, чем макрофагов. Такая этапность свойственна любому воспалительному процессу, что подчеркивает идентичность процессов, протекающих при воспалении жировой ткани с классическим воспалительным процессом, направленным на борьбу, например, с инфекцией [3][4][5][6][8][9]. По данным ряда авторов, ожирение сопровождается инфильтрацией жировой ткани макрофагами, с прямо пропорциональным количеством накопления этих клеток, количеству жировой ткани. При достижении больших степеней ожирения количество макрофагов в жировой ткани может достигать от 40 до 50% всех клеток [6][7][10][11][12]. Также отмечено, что инфильтрация макрофагами висцеральной части жировой ткани, превалирует над инфильтрацией в подкожном жире [3][4][5][13][14]. Кроме того, с прогрессией накопления жировой ткани прогрессивно увеличивается накопление и макрофагов [3][4][5][15]. Иначе говоря, процесс накопления жировой ткани сопровождается повышением выраженности процесса воспаления. В то же время установлено, что снижение объема жировой ткани сопровождается снижением инфильтрации жировой ткани макрофагами, то есть выраженностью воспаления [3][4][5][16][17]. Есть у макрофагов, рекрутированных в жировую ткань, особая тропность к гипертрофированным и погибающим адипоцитам располагаться вокруг них в виде венца [3][4][5][18]. Как правило это макрофаги класса М1, то есть имеющие провоспалительную направленность [3][4][5][18]. Ряд ученых высказал предположение о том, что рекрутирование макрофагов связано с неминуемым апоптозом гипертрофированных клеток [19]. Половина этих макрофагов погибает [18]. Кроме макрофагов М1 существуют макрофаги М2. Если первые являются провоспалительными, то вторые, наоборот, обладают противовоспалительной направленностью. В жировой ткани обнаруживаются и те, и другие, но при наборе веса, как правило, баланс смещен в пользу М1, что подчёркивает прогрессию воспаления [11][20[]21]. Макрофагам присваивают одно из важнейших защитных свойств — препятствие в преобразовании преадипоцитов в зрелые адипоциты [22]. То есть таким образом организм блокирует развитие гиперплазии жировой ткани, и избыточно поступающие энергетические ресурсы продолжают накапливаться в имеющихся адипоцитах, способствуя их дальнейшей гипертрофии. Преадипоциты, которым заблокирован путь преобразования в адипоциты, приобретают характеристики стромальных клеток жировой ткани и продуцируют элементы экстрацеллюлярного матрикса [15], что приведет к развитию фиброза. Еще одно, не менее важное свойство, которое характерно макрофагам, это повышение экспрессии фактора ангиогенеза PDGF (platalet-derived growth factor), который является дирижёром в процессе формирования эндотелиальных клеток [23].
Итак, начало воспаления жировой ткани можно обозначить началом рекрутирования моноцитов и их экстравазацию из кровеносных сосудов в жировую ткань. После выхода из сосудистого русла моноциты приобретают статус макрофагов. Выход этих клеток из сосудов происходит на уровне капилляров и протекает в несколько этапов: связывание с молекулами адгезии; роллинг (перекатывание); активация; прикрепление моноцитов к сосудистой стенке; и собственно процесс выхода клетки за пределы сосуда [24] (рис. 4).

Самым важным моментом в этом процессе, без которого не произойдет взаимодействия лейкоцитов и эндотелия, является появление молекул клеточной адгезии на поверхности и эндотелиальных клеток и лейкоцитов [25]. В качестве первичных активаторов моноцитов выступают белковые молекулы семейства селектинов (Р- и Е-селектины), после чего возможен их роллинг к эндотелию, связывание с другими молекулами, ассоциированными с эндотелием — ICAM (intercellular adhesion molecule) и VCAM (vascular cell adhesion molecule). Затем путем взаимодействия с молекулами РЕСАМ-1 (platelet/ endothelial cell adhesion molecule-1), располагающимися на боковых поверхностях эндотелиальных клеток, увеличиваясь от сосудов к тканям, то есть с увеличением градиента концентрации этих молекул, что, собственно, и обеспечивает экстравазацию моноцитов. Отмечен факт увеличения количества перечисленных выше молекул у людей с избыточным количеством жировой ткани [26]. Что является пусковым крючком для начала рекрутирования моноцитов? За адгезию и миграцию лейкоцитов отвечают, в том числе, и хемокины. Хемокины в огромном количестве продуцируются адипоцитами, и существует много их разных видов [27]. Один из самых известных и активных вариантов хемокинов это МСР-1 (monocyte chemoattractant protein 1), или CCL2 (chemokine ligand-2). Всё было вполне логично и понятно до тех пор, пока не было обнаружено, что хемокины синтезируются не только гипертрофированными адипоцитами, но и макрофагами, расположенными рядом с адипоцитами, преимущественно М1 [3][4][5][25][26]. Налицо формирование порочного круга: чем больше макрофагов, тем больше синтез хемокинов, которые привлекут еще больше моноцитов, которые после экстравазации преобразуются в макрофаги, заблокируют до определенной степени преадипоциты, увеличивая гипертрофию адипоцитов, также секретирующих хемокины и их рецепторы, поддерживая воспаление. Вся система воспаления состоит из множества порочных кругов, возможно, именно поэтому из нее так сложно вырваться.
Становится очевидным, что вовлечение микроциркуляции является неотъемлемой частью этого процесса. Обнаружено, что кровоток в жировой ткани при её избытке, то есть при ожирении, уменьшается и замедляется, но не меняется реология крови [15][28][29]. Значит основная проблема, связанная с микроциркуляцией при ожирении обусловлена изменениями эндотелия, что подтверждается усилением проницаемости сосудистой стенки при росте объема жировой ткани за счет увеличения фенестраций капилляров [15]. Само по себе выражение «нарушение микроциркуляции» — это общее понятие, подразумевающее под собой огромный симптомокомплекс, состоящий из ряда патологических явлений. А если сказать, что замедление кровотока — это фактор, способствующий развитию ишемии, следствием чего будет развитие нарушение метаболизма жировой ткани, гипоксия и повышение риска сердечно сосудистых катастроф, то картина проясняется и замедление кровотока воспринимается совершенно иначе [30]. Гипоксия способствует синтезу и высвобождению каскада цитокинов, блокировке дифференцировки преадипоцитов в адипоциты и активному участию макрофагов с их синтезом цитокинов. Гипоксия проявляется не только этими явлениями, но и маркерами гипоксии, такими как транскрипционный фактор или фактор-1 альфа [3][4][5]. Интересен факт изменения метаболизма адипоцитов в условиях гипоксии, когда повышалась продукция GLUT4 (Glucose transporter type 4), соответственно транспортировка и утилизация глюкозы адипоцитами. Становится очевидным вывод: гипоксия жировой ткани поддерживает и стимулирует развитие воспаления и таких следствий воспаления, как атеросклероз или развитие сахарного диабета 2 типа [31]. В ряде исследований показана прямая связь между количеством инфильтрирующих жировую ткань макрофагов и выраженностью нарушения дилатации сосудов, зависящей от состояния эндотелия. Всё то, что происходит при воспалении жировой ткани, способствует патологии эндотелия. Например, повышенная секреция ингибитора активатора плазминогена-1 (ИАП-1), повышенный синтез факторов воспаления и свободных форм кислорода, способствующих окислительному стрессу, дислипидемия, повышенный уровень свободных жирных кислот, результат липогенеза de novo, то есть всё то, что приводит к дисфункции эндотелия, включая его гипертоническое ремоделирование и атеросклеротические изменения, с повышением сердечно-сосудистых рисков [15][32][33][34][35].
Жировая ткань — это полноценный эндокринный орган, синтезирующий огромное количество биологически активных субстанций, имеющих системное и локальное воздействие. При воспалении жировой ткани происходят активные изменения в синтезе этих молекул. Для начала необходимо описать некоторые из них, наиболее изученные и интересные. Начать хочется с фактора некроза опухолей альфа ФНО-α, одного из самых известных представителей цитокинов. Преимущественный синтез ФНО-α, жировой ткани, происходит не в адипоцитах, что казалось логичным, а в макрофагах [36]. Это один из самых многоликих факторов воспаления, принимающий активнейшее участие в развитие инсулинорезистентности (см. рис. 5).

Как же развивается процесс инсулинорезистентности? Понимание этого процесса принципиально важно для любого специалиста, занимающегося лечением людей с избыточной массой тела, даже если они не жалуются на ожирение и пришли на прием с совершенно другими проблемами. В нормальных условиях инсулин оказывает своё воздействие через инсулиновый рецептор, находящийся на клеточной мембране (рис. 5) [1]. В момент взаимодействия инсулина с его рецептором на внутренней стороне мембраны на участке инсулинового рецептора запускается процесс активации фермента тирозинкиназы, который стимулирует аутофосфорилирование тирозина, находящегося там же. Внутриклеточные адаптопротеины, прежде всего субстрат инсулинового рецептора (СИР-1 (IRS-1)) присоединяется к аутофосфорилированным участкам инсулинового рецептора, на нём запускается фосфорилирование тирозина. А это стимулирует регуляторные внутриклеточные сигнальные пути инсулина, что в итоге приведёт к синтезу белка GLUT-4 и обеспечит проникновение глюкозы в клетку. При развитии воспаления жировой ткани внутриклеточные сигнальные пути инсулина нарушаются в результате разных процессов, но следствием является одно и тоже — инсулинорезистентность. Например, повышенные уровни ФНО-α и ИЛ-6 также могут активировать инсулиновый рецептор, но в отличие от физиологического пути при активации инсулином своего рецептора и фосфорилировании тирозина, цитокины активируют серинкиназу, соответственно, запускается фосфорилирование другой аминокислоты — серина, в том числе и в СИР-1 [37]. Этот процесс инактивирует СИР-1 или приводит к его разрушению, блокируя фосфорилирование тирозина и в рецепторе инсулина и в СИР-1, в результате чего нарушается внутриклеточный сигнальный путь инсулина и характерные для инсулина действия. Иначе говоря, так развивается инсулинорезистентность [38]. ФНО-α стимулирует развитие инсулинорезистентности не только таким образом. Он способствует увеличению в крови свободных жирных кислот, и это еще один путь для развития инсулинорезистентности, причем во многих тканях [39]. Кроме того, ФНО-α подавляет активность генов, при стимуляции которых повышается депонирование и усвоение свободных жирных кислот и глюкозы, и наоборот, повышает активность генов, стимулирующих липогенез, подавляя секрецию адипонектина и стимулируя синтез ИЛ-6 [40]. Кроме того, он подавляет активность процесса оксидации жирных кислот, повышая экспрессию генов, контролирующих синтез холестерина и жирных кислот [40].
Во-первых, он активно стимулирует в клетках синтез не только себя самого и своих же рецепторов, но и синтез других факторов воспаления. Во-вторых, он поддерживает продолжение воспалительной инфильтрации тканей, повышая экспрессию молекул адгезии на мембранах клеток. В-третьих, он выступает индуктором метаболизма арахидоновой кислоты, а это будет повышать степень выраженности воспаления, синтез простагландинов серии 2 и 4, тромбоксана, нарушая процессы свертывания крови и повышая риск тромбообразования. В-четвертых, он также активно стимулирует синтез факторов хемотаксиса и прежде всего МСР-1, что поддерживает и усиливает дальнейшую инфильтрацию жировой ткани [41]. В-пятых, помня о том, что синтез оксида азота — это инсулинозависимый процесс, ФНО-α способствует поддержанию и индукции патологии эндотелия, через угнетение дилатации сосудистой стенки посредством снижения синтеза оксида азота [41]. По данным ряда исследований, подтверждена четкая взаимосвязь между уровнями в крови интерлейкина-6 и ФНО-α с состоянием эндотелия у пациентов, имеющих в анамнезе гипертоническую болезнь [2]. В-шестых, высказывается гипотеза о том, что именно ФНО-α активно влияет на способность макрофагов блокировать преобразование преадипоцитов в адипоциты, а соответственно, очевидны два результата из этого явления — стимуляция фиброза жировой ткани и прогрессия ожирения. Седьмая функция, которая очевидно связана с ФНО-α-это активация внутриклеточных воспалительных сигнальных путей, в том числе и IκB (Inhibitor of kappa B), IKKβ (IκB Kinaseβ), NF-κB (Nuclear Factor-kappa B), с-Jun N-терминальную киназы (JNK), что повышает синтез факторов воспаления и реактивных форм кислорода, а это в свою очередь поддерживает и стимулирует воспаление [3][4][5]. Опять же очередной порочный круг, так свойственный системе воспаления. В-восьмых, ФНО-α ингибирует активность промотора гена адипокина, который обладает выраженными противовоспалительными свойствами адинонектина [2]. В-девятых, он же снижает экспрессию GLUT4 в клетках жировой и мышечной тканей [2], то есть основных потребителей глюкозы, запуская тем самым через ряд метаболических действий глюконеогенез в печени. В-десятых, в клетках печени ФНО-α повышает экспрессию генов, участвующих в производстве жирных кислот, и подавляет экспрессию тех генов, которые способствуют окислению жирных кислот. Иначе говоря, стимулируется синтез липопротеидов очень низкой плотности и гипертриглицеридемия [2]. Нельзя однозначно, негативно или позитивно, расценивать любую молекулу в организме. Тот же ФНО-α, который вызывает инсулинорезистентность в организме беременных женщин, способствуя перераспределению энергетических ресурсов от матери к плоду, обеспечивая таким образом рост плода, воспроизводство в частности и сохранение жизни, в целом. Синтез ФНО-α у беременных женщин обеспечивается плацентой, особенно повышено продуцируя этот цитокин во второй половине беременности. При этом около 94 % ФНО-α, продуцируемого плацентой, выбрасывается в кровь матери и только 6 % — в кровоток плода [7]. Здесь уместно напомнить, что инсулинорезистентность бывает и физиологической, а не только патологической, как в случае с беременностью или с пубертатным периодом у подростков, обеспечивая рост тела.
Не менее популярным, но имеющим противоположные свойства, является адипонектин (GBP28, apM1, AdipoQ и Acrp30). Его ген ACDC (adipocyte gene, C1q and collagen domain containing) расположен на хромосоме 3q27 на локусе, который ассоциируется с развитием метаболического синдрома на фоне висцеральной формы ожирения. Этот адипокин вырабатывается примерно в 100 раз больше, чем другие адипокины, и составляет у мужчин около 8 – 30 мкг мл [3][4][5]. Его главный производитель — это адипоциты, причем, по мнению ряда ученых, преимущественно клетки, находящиеся в подкожной части жировой ткани (табл. 1) [2] и плацентой, во время беременности. Но существуют и прямо противоположные представления о месте синтеза этого адипокина.
Таблица / Table 1
Факторы высвобождаемые и экспрессируемые разными отделами жировой ткани-подкожной и висцеральной, с акцентом, на количество тех или иных адипокинов, в разных частях [2]
Factors released and expressed by different parts of adipose tissue, subcutaneous and visceral, with an emphasis on the amount of certain adipokines, in different parts [2]


Если посмотреть на данные о подкожной жировой ткани в таблице, то видно, что именно здесь в большем количестве синтезируются противовоспалительные адипокины, такие как ASP, адипонектин, лептин и PPAR-γ. И именно поэтому с высокой долей вероятности у людей с метаболически здоровым ожирением преобладает подкожная часть жировой ткани над висцеральной.
Основными функциями противовоспалительного фактора адипонектина является подавление выраженности воспаления через ингибирующее действие транскрипционного фактора NF-κB, регуляция внутриклеточного уровня триглицеридов, снижение гипергликемии, балансирование энергетического гомеостаза и антиатерогенное действие, что в комплексе способствует снижению выраженности инсулинорезистентности и воспаления [3][5][42][43]. Адипонектин всегда будет снижен при высокой степени выраженности воспаления жировой ткани. По результатам инструментальных методов исследования содержание адипонектина обеспечивается синтезом абдоминально расположенной жировой ткани, и чем выше ИМТ и соотношение окружность талии/ окружность бедер, тем меньше уровень адипонектина [44][45]. Кроме того, синтез этого адипокина не зависит от таких факторов, как возраст, прием пищи или краткосрочный период голода и суточные ритмы [37]. Этот адипокин снижает секрецию провоспалительных цитокинов и не только ФНО-α, но и ИЛ-6, ПКС, но и факторов хемотаксиса [46]. По мнению некоторых авторов, существует обратная корреляция между уровнем адипонектина и СРБ [47]. Четкого понимания причин снижения адипонектина пока нет. Можно предположить, что наличие полиморфизма гена адинонектина (ADIPOQ), будет способствовать развитию, инсулинорезистентности, метаболического синдрома и прочим патологиям, сопровождающимся субклиническим воспалением жировой ткани. Противоатерогенное действие адипонектина объясняется его способностью связываться с субэндотелиально расположенным коллагеном в тех местах, где возникает поражение сосудистой стенки. Под воздействием адипонектина подавляется активность транскрипционного фактора NF-κB, экспрессия молекул адгезии VCAM, ICAM-1 и Е-селектина, пролиферация гладкомышечных клеток и дисфункция эндотелия [2]. Экспериментальные работы по введению рекомбинантного адипонектина крысам с индуцированным атеросклерозом продемонстрировали положительный эффект: адипонектин вызывал существенное улучшение состояние эндотелия и останавливал прогрессирование атеросклероза. Длительные наблюдения за индейцами племени Пима продемонстрировали обратную зависимость между гипоадипонектинемии и выраженностью инсулинорезистентности/гиперинсулинемии и вероятностью развития сахарногодиабета 2 типа. А повышение уровня адипонектина в крови уменьшает риск заболеваемости диабетом 2 типа, независимо от других факторов [48][49][50].
Один из наиболее изученных и часто упоминаемых цитокинов- интерлейкин-6 - ИЛ-6. Этот цитокин обладает выраженной провоспалительной направленностью, за которым закрепился статус одного из «организаторов» инсулинорезистентности, активно синтезируемый макрофагами, преимущественно абдоминальной частью жировой ткани, эндотелиоцитами в период воспаления, гипоксии, при травмах [37]. Но возникающая под его воздействием инсулинорезистентность обладает принципиальной избирательностью. То есть в жировой и печеночной тканях действительно возникает инсулинорезистентность, а вот в нервной или мышечной тканях, ИЛ-6, наоборот, повышается чувствительность этих тканей к инсулину. Его концентрация находится в положительной зависимости от величины ИМТ и, по ряду данных, от величины комплекса интима-медиа, и прогрессивно растет при развитии инсулинорезистентности и сахарном диабете 2 типа. Но в человеческом организме нет ничего однозначного. Так, и ИЛ-6 обладает и противоположными свойствами. Например, он способен уменьшить выраженность воспаления путем подавления синтеза ряда цитокинов, в том числе и ФНО-α [37]. ИЛ-6, как ФНО-α, обладают аутокринным и паракринным воздействием, и их концентрация в тканях в 100 раз выше, чем в крови, ввиду чего лабораторная диагностика анализов крови на уровень этих цитокинов не обладает объективностью, которая достоверно отображала бы происходящее в организме [37]. Интересные экспериментальные данные были представлены в конце прошлого века, когда в мозг животным вводили ИЛ-6 и это приводило к снижению веса через повышение расхода энергии. Эта же информация согласуется с тем, что уровень ИЛ-6 именно в центральной нервной системе находится в отрицательной корреляции с весом человека. При этом, мыши с полиморфизмом гена, ответственного за синтез ИЛ-6 и высоким его уровнем, имеют нарушения роста, низкий вес и уменьшение содержания жира [51]. Исходя из этого можно сделать вывод о неоднозначности и разнонаправленности воздействия на энергетический баланс ИЛ-6 в разных системах организма. Кроме нервной ткани мышечная ткань относится к тем, на которые ИЛ-6 оказывает больше положительное влияние, чем отрицательное. Достаточно давно в экспериментальных работах было продемонстрировано, что ИЛ-6 способствует утилизации глюкозы мышечными тканями, то есть действует физиологично [52]. Показано также, что умеренная физическая активность существенно повышает секрецию ИЛ-6 мышечными клетками, способствуя восстановлению чувствительности к инсулину и предотвращению больших метаболических патологий, в очередной раз подтверждая полезность физической активности [52][53][54]. При этом синтез других провоспалительных цитокинов не повышается. Очень важным аспектом, определяющим повышение синтеза ИЛ-6, является количество гликогена в миоцитах и обратно-пропорциональная зависимость синтеза ИЛ-6 от этого количества [55][56][57]. При проведении исследования на здоровых добровольцах, проводя инфузию ИЛ-6 в течение трех часов, отметили, что повышался липолиз, оксидация жиров, при этом не изменялась концентрация катехоламинов, инсулина или глюкагона [57]. Иначе говоря, ИЛ-6 является неким «надзирателем» над энергетическими ресурсами мышечной ткани и активно синтезируется при повышении физической активности, помогая высвобождать энергетические ресурсы-глюкозу, свободные жирные кислоты, и в то же время способствует их «усвоению» мышечными клетками. В спортивной медицине стабилизацию его уровня при регулярных тренировках называют «эффектом тренированности» [37]. В качестве примера — данные, которые приводил в своей статье профессор Шварц В., о том, что у здоровых людей ИЛ-6 в крови в среднем составляет от 1 до 2 пг/мл. После физической активности его уровень повышается через 1–3 часа. Отмечается зависимость уровня ИЛ-6 от интенсивности физической нагрузки. При умеренной нагрузке (около 40 % максимального потребления О2-V02 Max) у велосипедиста уровень ИЛ-6 существенно не менялся, но при повышении нагрузки при езде на велосипеде (60 % максимального потребления О2) уровень ИЛ-6 через три часа был на уровне 25 пг/мл [1]. А при сверхнагрузках, например, при марафонском забеге, уровень ИЛ-6 достигал 80 пг/мл.
Не менее важную роль ИЛ-6 играет и в работе иммунной системы, активизируя ее, в том числе и в аспекте дифференцировки Т-клеток, вызревании В-клеток, стимуляции синтеза гемопоэза, синтезе С-реактивного белка, выступая в роли двуликого Януса, будучи про- и противовоспалительным маркером системы воспаления [37].
Еще одним достаточно хорошо изученным адипокином является лептин, на который возлагались большие надежды ученых и врачей. Но все оказалось не так просто. Лептин был выделен сравнительно недавно, в 1994 г. Синтезируется этот адипокин, преимущественно, адипоцитами. Вместе с инсулином, лептин относится к тем соединениям, которые отражают объем жировой ткани [58]. Лептин еще называют гормоном насыщения, потому что он снижает аппетит и стимулирует прекращение приема пищи, воздействуя на центры в дугообразных ядрах гипоталамуса. В экспериментальных работах с животными, имеющими ожирение, экзогенный ввод лептина не приводил к положительным результатам ввиду развития лептинрезистентности по аналогии с инсулинорезистентностью, когда уровень гормона растет, а его эффекты не прослеживаются. Как и другие адипокины, несмотря на свой противовоспалительный профиль, лептин способен стимулировать и лейкоциты, макрофаги (рекрутированные моноциты) и лимфоциты к синтезу цитокинов [59]. Иначе говоря, противовоспалительный статус лептина не оправдался, и этот цитокин, как и большинство других, имеет разнонаправленное действие. Есть еще одно наблюдение на уровне гипотезы, пока не получившее достоверных доказательств: высокие уровни лептина способствуют повышению риска тромбообразования за счет воздействия на специфические рецепторы к лептину, расположенные на тромбоцитах.
Чем больше получаем информации о действии тех же цитокинов, тем больше имеем представление о глобальности их действия. Кроме адипокинов, то есть веществ, вырабатываемых в жировой ткани, есть другие ткани и вещества, вырабатываемые в них и оказывающие влияние на весь организм, включая жировую ткань. Например, белок, вырабатываемый остеобластами, то есть в костной ткани, остеокальцин, наиболее известный как маркер остеогенеза, играет еще очень много важнейших ролей в организме. Он — активный участник энергетической регуляции, так как повышает чувствительность к инсулину и способствует утилизации глюкозы. Кроме того, он стимулирует синтез адипонектина в клетках жировой ткани. Иначе говоря, налицо явно противовоспалительный акцент этого белка, который предложено считать остеокином по принципу названия продуцирующей ткани [60]. Но это — ещё не все известные его свойства. Он способствует повышению синтеза тестостерона, то есть обладает и анаболическим действием в отношении мышечной ткани [60]. У тестостерона широкий спектр воздействия на организм. Изучение мышечной ткани с её эндокринными свойствами привело к тому, что был предложен ещё один новый термин — адипомиокины, из названия которого становится ясно, что существуют
вещества, одинаково активные в отношении и жировой и мышечной, тканей и способные производится ими обеими. Один из наиболее хорошо изученных провоспалительных цитокинов ИЛ-6, признан и миокином, причем с явным противовоспалительным действием, направленным на повышение утилизации глюкозы мышцами (описан его эффект «тренированности», выше). Кроме ИЛ-6, ИЛ-7 и 8 повышают β-оксидацию жирных кислот, а ИЛ-15, во время активной мышечной работы, повышает утилизацию глюкоза [60]. То есть ученые подтверждают воздействие адипокинов на мышечную ткань и подчеркивают взаимосвязь и взаимодействие разных тканей организма.
Одним из звеньев воспаления жировой ткани на фоне возникшей инсулинорезистентности является активированный липолиз и липогенез de novo. Основным блокатором липолиза является инсулин, и при развитии инсулинорезистентности этот процесс становится бесконтрольным. Освобождающиеся в большом количестве свободные жирные кислоты являются потенциальными активаторами внутриклеточных провоспалительных киназ, таких как JNK (c-Jun N-terminale Kinase), IKKβ, протеинкиназа-С (ПК-С), что приводит к активации транскрипционного фактора NF-κB с запуском или поддержанием воспаления [1]. Кроме того, свободные жирные кислоты являются превосходным субстратом для производства на мультиферментном комплексе — синтазе жирных кислот [FASN, fatty acid synthase] [61]. Ее роль заключается в последовательном удлинении радикала жирных кислот на 2 углеродных атома, вплоть до получения 16-й углеродной насыщенной жирной кислоты — пальмитиновой. Донором 2-х углеродных атомов является малонил-КоА. Избыток пальмитиновой кислоты является потенциальным стимулятором TLR4 (Toll-подобный рецептор 4), которые также способны напрямую активировать транскрипционный фактор NF-κB и провоспалительные киназы, стимулирующие развитие воспаления или его поддержание и усиление [2].
Оксидативный стресс — это нарушенный в пользу последних баланс между восстановителями и окислителями. Повышенный уровень липидов, прежде всего, в адипоцитах, потенцирует НАДФ-оксидазу (никотинамидадениндинуклеотидфосфат оксидаза) [1][2], которая локализуется, преимущественно, на мембранах митохондрий. Это повышает количество оксидантов и, прежде всего, супреоксида, который синтезируется, в основном, в макрофагах2, но не только. Его синтез налажен и в нейтрофилах, в гладкомышечных и эндотелиальных клетках, также в фибробластах [62]. Это ведет к снижению уровня суперокиддисмутазы (СОД), одного из самых мощных антиоксидантов, и синтезу внутриклеточных киназ: протеинкиназ С (β, σ, θ), JNK, p38 MARK, IKKβ. Может активироваться синтез керамидов. Всё это в комплексе, накапливаясь, например, в миоцитах, способствуют активации всё того же фактора транскрипции NF-κB, с дальнейшим синтезом цитокинов и реактивных форм кислорода, с развитием или усилением воспаления, его порочных кругов и ещё большему накоплению жировой ткани [2]. В ответ на воспалительную реакцию повышается синтез окиси азота, который при низком уровне СОД и высоком уровне супероксида, вступая в реакцию с ним, стимулирует синтез группы одних из самых мощных оксидантов — пероксинитратов. Они в свою очередь блокируют активность фермента цис-аконитазы в цикле трикарбоновых кислот и комплекс II в реакции окислительного фосфорилирования. Результатом подобных действий является разобщение окислительного фосфорилирования, повреждение ДНК и прогрессирование воспаления (рис. 2), источник указан в сноске 1.
Врачей всегда интересует практическое выражение научных изысканий и возможное практическое применение этих знаний. Например, как воспаление может воздействовать на гормональный фон у мужчин? Очень интересные данные представили ученые в 2004 г. [63], когда провели исследование мочи на наличие и соотношение метаболитов эстрогенов гидроксилированных форм 2-гидроксиэстрона (2-(ОН)-Е1) и 16–гидроксиэстрона (16-(ОН)-Е1) у пациентов с воспалительными заболеваниями, такими как ревматоидный артрит и системная красная волчанка. Результаты были таковыми: у пациентов с данными заболеваниями 2-(ОН)-Е1 был в 10 раз ниже, чем у здоровых пациентов, и соотношение 16-(ОН)-Е1/2-(ОН)-Е1 было в 20 раз выше, чем в контрольной группе. Напомним, что 2-(ОН)-Е1 — это слабый в аспекте эстрогенного действия метаболит, не несущий никаких угроз человеку, получающий статус онкопротекторного после прохождения стадии метоксилирования. В тот же момент 16-(ОН)-Е1 — это сильный метаболит с выраженной эстрогенной активностью, обладающий существенным митогенным воздействием на клетки. Ученые сделали вывод, что воспалительный процесс повышает вероятность метаболизма эстрогенов по пути синтеза 16-(ОН)-Е1, а он в свою очередь поддерживает дальнейшее воспаление и пролиферацию, свойственную этим заболеваниям. Кроме того, ученые продемонстрировали существенное преобладание эстрогенов над уровнем андрогенов в синовиальной жидкости таких пациентов. Еще больший интерес вызывает информация о том, что в инфильтрированных макрофагами тканях была выявлена положительная корреляция между активностью ароматазы и синтезом цитокинов. То есть повышенное производство факторов воспаления, таких как ФНО-α, ИЛ-1 или ИЛ-6, повышают активность ароматазы, а она способствует избыточной конвертации тестостерона в эстрадиол с развитием андрогенного дефицита и гиперэстрогении, способствующей дальнейшей активации транскрипционного фактора воспаления -NF-κB. То есть налицо очередной порочный круг. Тестостерон, по мнению автора (и мы с этим согласны), вызывает проапоптический и подавляющий эффект в отношении макрофагов и прямо противоположный эффект с эстрадиолом. Иначе говоря, оба стероидных гормона действуют на процесс воспаления с диаметрально противоположным эффектом [63].
Таким образом, иллюзии о том, что ожирение — это косметический дефект, с которым легко справиться усилием воли или рядом хирургических операций, начиная от липосакции и заканчивая бариатрическими операциями, утрачены. Результаты множества исследований не оставили сомнений в том, что избыток жировой ткани стимулирует развитие ожирения. Именно избыточно калорийная пища и сниженная физическая активность индуцируют сначала накопление жировой ткани, в которой и развивается субклиническое воспаление. С учетом того, что жировая ткань иногда составляет более 50 % от всей массы тела, масштаб возникающих проблем сложно переоценить. Весь процесс воспаления сопровождается формированием порочных кругов, когда цитокины стимулируют синтез оксидантов, а те поддерживают и стимулируют синтез цитокинов; когда цитокины стимулируют не только синтез других цитокинов, но и самих себя и свои же рецепторы; когда избыточно развитые адипоциты стимулируют синтез хемокинов, те привлекают моноциты, они, став макрофагами, также стимулируют цитокины и хемокины, способствуя дальнейшему росту ожирения и синтезу цитокинов; или когда цитокины повышают активность ароматазы, та, снижая уровень тестостерона и повышая уровень эстрадиола, сокращая объем мышечной ткани и физической активности, способствует росту ожирения, дальнейшей активации ароматазы, и повышению уровня гиперэстрогении, которая в свою очередь усиливает синтез цитокинов, повышая активность внутриклеточных провоспалительных киназ [63]. Подобных порочных кругов в системе воспаления множество. Эта одна из основ воспаления. Иначе говоря, однажды начавшись, процесс воспаления жировой ткани становится самоподдерживающимся процессом, усиливая и воспаление и стимулируя дальнейший рост жировой ткани. Этот рост жировой ткани, а в дальнейшем и её отложение в мышечной, печеночной и других тканях, обусловлен в последующем не только поступлением избытка жиров с пищей, но и включением в процесс повышенного липолиза на фоне инсулинорезистентности и снижения уровня оксидации жиров за счет блокады PPARγ и АМФК (аденозин монофосфат-активируемая протеинкиназа) и уровня адипонектина. При множестве эффективных средств, в том числе и медикаментозных, самым лучшим и надежным средством, особенно в совокупности с использованием лекарственных средств, является модификация образа жизни. Этому важнейшему разделу мы посвятим отдельную статью. И конечно же, несмотря на глобальный рост ожирения в мире, вернее, ориентируясь на этот глобальным рост ожирения, надо неустанно говорить о профилактике и ожирения, и воспаления этой ткани, потому что врачебным искусством и является недопущение развития заболевания.
Вместе с ростом количества людей с избыточной массой тела и / или ожирением в масштабе эпидемии прогрессирует и рост онкологических заболеваний. Несмотря на то, что ожирение в 95 % случаев — это модифицируемый фактор, большинство людей самостоятельно не могут справиться с этим недугом, а значит коморбидные патологии развиваются также масштабно, как и само ожирение, включая рак предстательной железы (РПЖ), который является одним из самых распространенных раков среди мужского населения. Долгое время основной причиной РПЖ считался тестостерон. Рассматривать ожирение как потенциальную причину этого или другого онкологического процесса никому не приходило в голову. Этот интерес возник значительно позже (рис. 6).
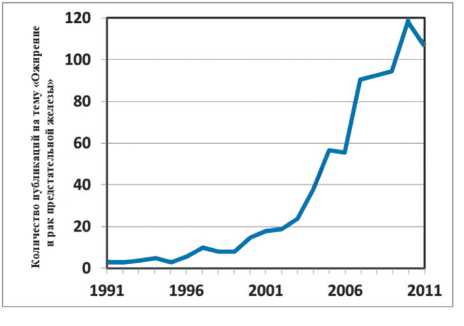
В 2013 г. были опубликованы результаты метаанализа, который включил в себя статьи с 1991 по 2012 гг. [64]. Данные, которые интересовали ученых, были следующими: наличие ожирения, ИМТ, риск развития рака простаты, заболеваемость раком простаты, смертность от рака простаты, радикальная простатэктомия, андрогенная депривационная терапия, лучевая терапия, брахитерапия, рак простаты и качество жизни с этим заболеванием, рак простаты и активное наблюдение, инсулин, инсулиноподобный фактор роста, андроген, эстрадиол, лептин, адипонектин и ИЛ-6. Из полученных данных ученые сделали вывод о том, что ожирение связано в повышенной частотой возникновения агрессивной формы РПЖ; повышенным риском развития дефицита гормонов после радикальной простатэктомии и лучевой терапии с использованием внешнего воздействия (луча); более высокой частотой осложнений после проведения андроген-депривационной терапии и повышенной смертностью, специфичной для рака простаты. На животных моделях было показано, что снижение веса замедляло прогрессирование рака простаты. На рис. 7 показана взаимосвязь ожирения и механизмов развития рака простаты [64].
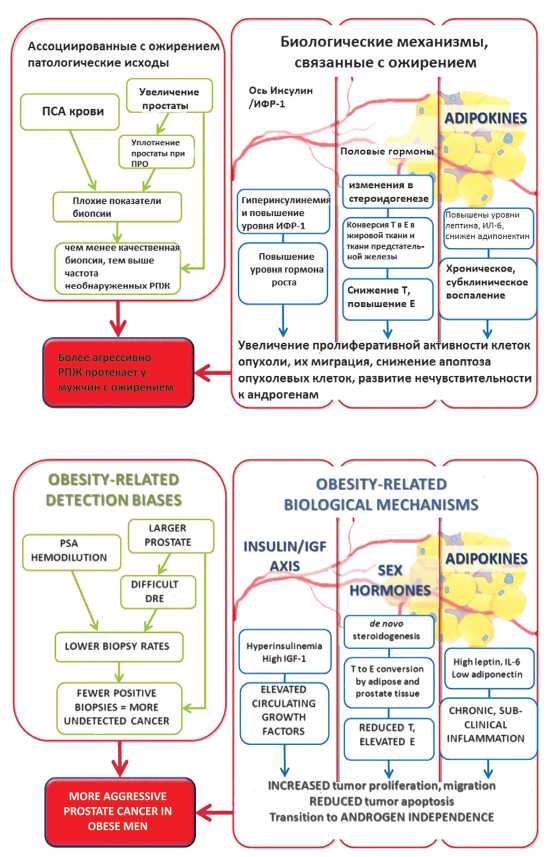
Таким образом, можно предположить, что ожирение стимулирует агрессивное течение рака простаты. Также исследователи предложили клинические рекомендации в виде алгоритма диагностики и лечения ожирения у мужчин с раком предстательной железы (см.рис.8) [64].

С учетом сложности и многофакторности патогенеза ожирения, в том числе и у мужчин, хотелось бы, чтобы в современных алгоритмах диагностики, а в последующем и в лечении мужчин с РПЖ, был бы обозначен не только контроль за органическими изменениями, например, объёма предстательной железы, но и лабораторные изменения уровня общего тестостерона, эстрадиола, инсулина, так как они играют ключевую роль в происходящих органических изменениях. И самое главное, приводя состав тела к норме, мы делаем свою практику более эффективной, потому что воздействуем на причину, а не на следствие. Кроме того, достигая физиологических параметровсостава тела, мы решаем сразу много задач, а не одну, так как избыточная масса тела и / или ожирение всегда сопровождаются большим количеством коморбидных патологий. Мы выражаем надежду на то, что наши научные работы внесут свой вклад в расширение границ осознания масштабности и сложности такого заболевания, как ожирение, и ключевого звена его патогенеза — воспаления.
Финансирование. Исследование не имело спонсорской поддержки.
Financing. The study did not have sponsorship.
Конфликт интересов. Авторы заявляют об отсутствии конфликта интересов.
Conflict of interest. Authors declares no conflict of interest.
1. Kharrazian, D. Заболевания желудочно-кишечного тракта: результаты последних исследований и и комментарии. Дата доступа — 02.04.2016; Доступно по адресу: http://www.applied-kinesiology.ru/book/107
2. Презентация-патофизиология воспаления. ФГБУ «СЗФМИЦ им. В.А. Алмазова» Минздрава России
1. Шварц В.Я. Воспаление как фактор патогенеза инсулинорезистентности и сахарного диабета 2-го типа // Терапевтический архив. - 2009. - № 10. - С.74-80. eLIBRARY ID: 13055602
2. Абатуров А.Е. Особенности метаболического синдрома у детей. // Дитячий лікар. - 2011. - № 4. - С.54-61.
3. Шварц В.Я. Воспаление жировой ткани // Проблемы эндокринологии. - 2009. - Т.55, № 4. - С.44-49. https://doi.org/10.14341/probl200955444-49
4. Шварц В.Я. Воспаление жировой ткани // Проблемы эндокринологии. - 2009. - Т.55, № 5. - С.43-48. https://doi.org/10.14341/probl200955543-48
5. Шварц В.Я. Воспаление жировой ткани // Проблемы эндокринологии. - 2009. - Т.55, № 6. - С.40-45. https://doi.org/10.14341/probl200955640-45
6. Шварц В.Я. Жировая ткань как эндокринный орган. // Проблемы эндокринологии. - 2009. - Т.55, №1. - С. 38-43. https://doi.org/10.14341/probl200955138-43
7. Шварцбурд П.М. Разные лица инсулиновой резистентности. // Химия и Жизнь. - 2013. - № 7. - С.2-5.
8. Iizuka K. The Role of Carbohydrate Response Element Binding Protein in Intestinal and Hepatic Fructose Metabolism. // Nutrients. - 2017. - V.9, № 2. – P. 181. https://doi.org/10.3390/nu9020181
9. Kintscher U., Hartge M., Hess K., Foryst-Ludwig A., Clemenz M., Wabitsch M., et al. T-lymphocyte infiltration in visceral adipose tissue: a primary event in adipose tissue inflammation and the development of obesity-mediated insulin resistance. // Arterioscler Thromb Vasc Biol. - 2008. - V.28, № 7. - P.1304-10. https://doi.org/10.1161/atvbaha.108.165100
10. Cancello R., Henegar C., Viguerie N., Taleb S., Poitou C., et al. Reduction of macrophage infiltration and chemoattractant gene expression changes in white adipose tissue of morbidly obese subjects after surgery-induced weight loss. // Diabetes. - 2005. - V.54, № 8. - P.2277-86. https://doi.org/10.2337/diabetes.54.8.2277
11. Huber J., Kiefer FW., Zeyda M., Ludvik B., Silberhumer GR., Prager G., et al. CC chemokine and CC chemokine receptor profiles in visceral and subcutaneous adipose tissue are altered in human obesity. // J Clin Endocrinol Metab. - 2008. - V.93, № 8. - P.3215-21. https://doi.org/10.1210/jc.2007-2630
12. Weisberg SP., McCann D., Desai M., Rosenbaum M., Leibel RL., Ferrante A.W. Jr. Obesity is associated with macrophage accumulation in adipose tissue. // J Clin Invest. - 2003. - V.112, № 12. - P.1796-808. https://doi.org/10.1172/jci19246
13. Cancello R., Clément K. Is obesity an inflammatory illness? Role of low-grade inflammation and macrophage infiltration in human white adipose tissue. // Bjog. - 2006. - V.113, № 10. - P.1141-7. https://doi.org/10.1111/j.1471-0528.2006.01004.x
14. Sell H., Dietze-Schroeder D., Eckel J. The adipocyte-myocyte axis in insulin resistance. // Trends Endocrinol Metab. - 2006. - V.17, № 10. - P.416-22. https://doi.org/10.1016/j.tem.2006.10.010
15. Nishimura S., Manabe I., Nagasaki M., Seo K., Yamashita H., et al. In vivo imaging in mice reveals local cell dynamics and inflammation in obese adipose tissue. // J Clin Invest. - 2008. - V.118, № 2. - P.710-21. https://doi.org/10.1172/jci33328
16. Clément K., Viguerie N., Poitou C., Carette C., Pelloux V., et al. Weight loss regulates inflammation-related genes in white adipose tissue of obese subjects. // Faseb j. - 2004. - V.18, № 14. - P.1657-69. https://doi.org/10.1096/fj.04-2204com
17. Cancello R., Tordjman J., Poitou C., Guilhem G., Bouillot J.L., et al. Increased infiltration of macrophages in omental adipose tissue is associated with marked hepatic lesions in morbid human obesity. // Diabetes. - 2006. - V.55, № 6. - P.1554- 61. https://doi.org/10.2337/db06-0133
18. Murano I., Barbatelli G., Parisani V., Latini C., Muzzonigro G., et al. Dead adipocytes, detected as crown-like structures, are prevalent in visceral fat depots of genetically obese mice. // J Lipid Res. - 2008. - V.49, № 7. - P.1562-8. https://doi.org/10.1194/jlr.M800019-JLR200
19. Cinti S., Mitchell G., Barbatelli G., Murano I., Ceresi E., et al. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. // J Lipid Res. - 2005. - V.46, № 11. - P.2347-55. doi: 10.1194/jlr.M500294-JLR200.
20. Bourlier V., Zakaroff-Girard A., Miranville A., De Barros S., Maumus M., et al. Remodeling phenotype of human subcutaneous adipose tissue macrophages. // Circulation. - 2008. - V.117, № 6. - P.806-15. doi: 10.1161/CIRCULATIONAHA.107.724096.
21. Lumeng CN., Bodzin JL., Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. // J Clin Invest. - 2007. - V.117, № 1. - P.175-84. DOI: 10.1172/jci29881
22. Constant VA., Gagnon A., Yarmo M., Sorisky A. The antiadipogenic effect of macrophage-conditioned medium depends on ERK1/2 activation. // Metabolism. - 2008. - V.57, № 4. - P.465-72. DOI: 10.1016/j.metabol.2007.11.005
23. Pang C., Gao Z., Yin J., Zhang J., Jia W., Ye J. Macrophage infiltration into adipose tissue may promote angiogenesis for adipose tissue remodeling in obesity. // Am J Physiol Endocrinol Metab. - 2008. - V.295, № 2. - P.E313-22. doi: 10.1152/ajpendo.90296.2008
24. Hatoum OA., Heidemann J., Binion DG. The intestinal microvasculature as a therapeutic target in inflammatory bowel disease. // Ann N Y Acad Sci. - 2006. - V.1072, № - P.78-97. doi: 10.1196/annals.1326.003
25. Blankenberg S., Barbaux S., Tiret L. Adhesion molecules and atherosclerosis. // Atherosclerosis. - 2003. - V.170, № 2. - P.191-203. doi: 10.1016/s0021-9150(03)00097-2
26. Miller MA., Cappuccio FP. Cellular adhesion molecules and their relationship with measures of obesity and metabolic syndrome in a multiethnic population. // Int J Obes (Lond). - 2006. - V.30, № 8. - P.1176-82. doi: 10.1038/sj.ijo.0803264
27. Kanda H., Tateya S., Tamori Y., Kotani K., Hiasa K., et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. // J Clin Invest. - 2006. - V.116, № 6. - P.1494-505. doi: 10.1172/JCI26498
28. Kampf C., Bodin B., Källskog Ö., Carlsson C., Jansson L. Marked Increase in White Adipose Tissue Blood Perfusion in the Type 2 Diabetic GK Rat. // Diabetes. - 2005. - V.54, № 9. - P.2620. doi: 10.2337/diabetes.54.9.2620
29. Simonsen L., Enevoldsen LH., Bülow J. Determination of adipose tissue blood flow with local 133Xe clearance. Evaluation of a new labelling technique. // Clin Physiol Funct Imaging. - 2003. - V.23, № 6. - P.320-3. doi: 10.1046/j.1475-0961.2003.00509.x
30. Summers LKM., Samra JS., Frayn KN. Impaired postprandial tissue regulation of blood flow in insulin resistance: a determinant of cardiovascular risk? // Atherosclerosis. - 1999. - V.147, № 1. - P.11-15. doi: 10.1016/s0021-9150(99)00172-0
31. Trayhurn P., Wang B.,Wood I.S. Hypoxia in adipose tissue: a basis for the dysregulation of tissue function in obesity? // Br J Nutr. - 2008. - V.100, № 2. - P.227-35. doi: 10.1017/S0007114508971282
32. Kita T., Kume N., Minami M., Hayashida K., Murayama T., et al. Role of oxidized LDL in atherosclerosis. // Ann N Y Acad Sci. - 2001. - V.947, № - P.199-205; discussion 205-6. doi: 10.1111/j.1749-6632.2001.tb03941.x
33. Matsuoka H. Endothelial dysfunction associated with oxidative stress in human. // Diabetes Res Clin Pract. - 2001. - V.54 Suppl 2, № - P.S65-72. doi: 10.1016/s0168-8227(01)00337-0
34. McIntyre M., Bohr DF., Dominiczak AF. Endothelial function in hypertension: the role of superoxide anion. // Hypertension. - 1999. - V.34, № 4 Pt 1. - P.539-45. doi: 10.1161/01.hyp.34.4.539
35. Steinberg H.O., Tarshoby M., Monestel R., Hook G., Cronin J., et al. Elevated circulating free fatty acid levels impair endothelium-dependent vasodilation. // J Clin Invest. - 1997. - V.100, № 5. - P.1230-9. DOI: 10.1172/jci119636
36. Hoch M., Eberle A.N., Peterli R., Peters T., Seboek D., et al. LPS induces interleukin-6 and interleukin-8 but not tumor necrosis factor-alpha in human adipocytes. // Cytokine. - 2008. - V.41, № 1. - P.29-37. DOI: 10.1016/j.cyto.2007.10.008
37. de Alvaro C., Teruel T., Hernandez R., Lorenzo M. Tumor necrosis factor alpha produces insulin resistance in skeletal muscle by activation of inhibitor kappaB kinase in a p38 MAPK-dependent manner. // J Biol Chem. - 2004. - V.279, № 17. - P.17070-8. DOI: 10.1074/jbc.M312021200
38. Granner D.K., O’Brien R.M. Molecular physiology and genetics of NIDDM. Importance of metabolic staging. // Diabetes Care. - 1992. - V.15, № 3. - P.369-95. DOI: 10.2337/diacare.15.3.369
39. Ruan H., Lodish H.F. Insulin resistance in adipose tissue: direct and indirect effects of tumor necrosis factor-alpha. // Cytokine Growth Factor Rev. - 2003. - V.14, № 5. - P.447-55. DOI: 10.1016/s1359-6101(03)00052-2
40. Ruan H., Miles P.D., Ladd C.M., Ross K., Golub T.R., et al. Profiling gene transcription in vivo reveals adipose tissue as an immediate target of tumor necrosis factor-alpha: implications for insulin resistance. // Diabetes. - 2002. - V.51, № 11. - P.3176-88. DOI: 10.2337/diabetes.51.11.3176
41. Zhu J., Yong W., Wu X., Yu Y., Lv J., et al. Anti-inflammatory effect of resveratrol on TNF-alpha-induced MCP-1 expression in adipocytes. // Biochem Biophys Res Commun. - 2008. - V.369, № 2. - P.471-7. DOI: 10.1016/j.bbrc.2008.02.034
42. Hattori Y., Nakano Y., Hattori S., Tomizawa A., Inukai K., Kasai K. High molecular weight adiponectin activates AMPK and suppresses cytokine-induced NF-kappaB activation in vascular endothelial cells. // FEBS Lett. - 2008. - V.582, № 12. - P.1719-24. DOI: 10.1016/j.febslet.2008.04.037
43. Ouchi N., Kihara S., Arita Y., Okamoto Y., Maeda K., et al. Adiponectin, an adipocyte-derived plasma protein, inhibits endothelial NF-kappaB signaling through a cAMP-dependent pathway. // Circulation. - 2000. - V.102, № 11. - P.1296-301. DOI: 10.1161/01.cir.102.11.1296
44. Staiger H., Tschritter O., Machann J., Thamer C., Fritsche A., et al. Relationship of serum adiponectin and leptin concentrations with body fat distribution in humans. // Obes Res. - 2003. - V.11, № 3. - P.368-72. DOI: 10.1038/oby.2003.48
45. Cnop M., Havel P.J., Utzschneider K.M., Carr D.B., Sinha M.K., et al. Relationship of adiponectin to body fat distribution, insulin sensitivity and plasma lipoproteins: evidence for independent roles of age and sex. // Diabetologia. - 2003. - V.46, № 4. - P.459-69. DOI: 10.1007/s00125-003-1074-z
46. Cheng K.H., Chu C.S., Lee K.T., Lin T.H., Hsieh C.C., et al. Adipocytokines and proinflammatory mediators from abdominal and epicardial adipose tissue in patients with coronary artery disease. // Int J Obes (Lond). - 2008. - V.32, № 2. - P.268-74. DOI: 10.1038/sj.ijo.0803726
47. Engeli S., Feldpausch M., Gorzelniak K., Hartwig F., Heintze U., et al. Association between adiponectin and mediators of inflammation in obese women. // Diabetes. - 2003. - V.52, № 4. - P.942-7. DOI: 10.2337/diabetes.52.4.942
48. Weyer C., Funahashi T., Tanaka S., Hotta K., Matsuzawa Y., et al. Hypoadiponectinemia in obesity and type 2 diabetes: close association with insulin resistance and hyperinsulinemia. // J Clin Endocrinol Metab. - 2001. - V.86, № 5. - P.1930-5. DOI: 10.1210/jcem.86.5.7463
49. Lindsay R.S., Funahashi T., Hanson R.L., Matsuzawa Y., Tanaka S., et al. Adiponectin and development of type 2 diabetes in the Pima Indian population. // Lancet. - 2002. - V.360, № 9326. - P.57-8. DOI: 10.1016/s0140-6736(02)09335-2
50. Spranger J., Kroke A., Möhlig M., Bergmann M.M., Ristow M., et al. Adiponectin and protection against type 2 diabetes mellitus. // Lancet. - 2003. - V.361, № 9353. - P.226-8. DOI: 10.1016/s0140-6736(03)12255-6
51. De Benedetti F., Alonzi T., Moretta A., Lazzaro D., Costa P., et al. Interleukin 6 causes growth impairment in transgenic mice through a decrease in insulin-like growth factor-I. A model for stunted growth in children with chronic inflammation. // J Clin Invest. - 1997. - V.99, № 4. - P.643-50. DOI: 10.1172/jci119207
52. Lee M.D., Zentella A., Vine W., Pekala P.H., Cerami A. Effect of endotoxin-induced monokines on glucose metabolism in the muscle cell line L6. // Proc Natl Acad Sci U S A. - 1987. - V.84, № 9. - P.2590-4. DOI: 10.1073/pnas.84.9.2590
53. Ostrowski K., Rohde T., Asp S., Schjerling P., Pedersen BK. Pro- and anti-inflammatory cytokine balance in strenuous exercise in humans. // J Physiol. - 1999. - V.515 ( Pt 1), № Pt 1. - P.287-91. DOI: 10.1111/j.1469-7793.1999.287ad.x
54. Febbraio M.A.,Pedersen B.K. Muscle-derived interleukin-6: mechanisms for activation and possible biological roles. // Faseb j. - 2002. - V.16, № 11. - P.1335-47. DOI: 10.1096/fj.01-0876rev
55. Steensberg A., Febbraio M.A., Osada T., Schjerling P., van Hall G., et al. Interleukin-6 production in contracting human skeletal muscle is influenced by pre-exercise muscle glycogen content. // J Physiol. - 2001. - V.537, № Pt 2. - P.633-9. DOI: 10.1111/j.1469-7793.2001.00633.x
56. Febbraio M.A., Steensberg A., Keller C., Starkie R.L., Nielsen H.B., et al. Glucose ingestion attenuates interleukin-6 release from contracting skeletal muscle in humans. // J Physiol. - 2003. - V.549, № Pt 2. - P.607-12. DOI: 10.1113/jphysiol.2003.042374
57. Hiscock N., Chan M.H., Bisucci T., Darby I.A., Febbraio M.A. Skeletal myocytes are a source of interleukin-6 mRNA expression and protein release during contraction: evidence of fiber type specificity. // Faseb j. - 2004. - V.18, № 9. - P.992- 4. DOI: 10.1096/fj.03-1259fje
58. Романцова Т.И. Молекулярные механизмы регуляции массы тела как мишени патогенетической терапии ожирения. // Терапия. - 2015. - Т.4, № 4. - С.71-78. eLIBRARY ID: 25512742
59. Fantuzzi G., Mazzone T. Adipose tissue and atherosclerosis: exploring the connection. // Arterioscler Thromb Vasc Biol. - 2007. - V.27, № 5. - P.996-1003. DOI: 10.1161/atvbaha.106.131755
60. Бородина С.В., Гаппарова К.М., Зайнудинов З.М., Григорьян О.Н. Генетические предикторы развития ожирения. // Ожирение и метаболизм. - 2016. - Т.13, № 2. - С.7-13. eLIBRARY ID: 27161173
61. Павлова З.Ш., Голодников И.И., Камалов А.А. Биохимические механизмы развития неалкогольной жировой болезни печени под воздействием фруктозы. // Технологии живых систем. - 2018. - Т.15, № 4. - С.18-27. DOI: 10.18127/j20700997-201804-02
62. Griendling KK., Sorescu D., Ushio-Fukai M. NAD(P)H oxidase: role in cardiovascular biology and disease. // Circ Res. - 2000. - V.86, № 5. - P.494-501. DOI: 10.1161/01.res.86.5.494
63. Cutolo M. Estrogen metabolites: increasing evidence for their role in rheumatoid arthritis and systemic lupus erythematosus. // J Rheumatol. - 2004. - V.31, № 3. - P.419-21. PMID: 14994382.
64. Allott E.H., Masko E.M., Freedland S.J. Obesity and prostate cancer: weighing the evidence. // Eur Urol. - 2013. - V.63, № 5. - P.800-9. DOI: 10.1016/j.eururo.2012.11.013
к.м.н., врач-эндокринолог, старший научный сотрудник отдела возрастассоциированных заболеваний,
Москва
врач-ординатор, кафедра эндокринологии,
Москва
Павлова З.Ш., Голодников И.И. Ожирение = воспаление. Патогенез. Чем это грозит мужчинам? Медицинский вестник Юга России. 2020;11(4):6-23. https://doi.org/10.21886/2219-8075-2020-11-4-6-23
Pavlova Z.Sh., Golodnikov I.I. Obesity = inflammation. Pathogenesis. How does this threaten men? Medical Herald of the South of Russia. 2020;11(4):6-23. (In Russ.) https://doi.org/10.21886/2219-8075-2020-11-4-6-23
344022, г. Ростов-на-Дону, пер. Нахичеванский, 29
Ростовский государственный медицинский университет
Тел.: +7 918 571 0558
E-mail: journal@medicalherald.ru


































