



Contents
Scroll to:
https://doi.org/10.21886/2219-8075-2021-12-1-24-32
Scroll to:
The efficacy of depressive disorders treatment is insufficient. It is explained by an incomplete understanding of both pathogenesis of depression and antidepressants mechanism action. An improvement of the treatment efficacy of depression disorders is closely associated with complete knowledge of the pathogenesis of disorders and antidepressant mechanism of action. The effect produced by the first line of antidepressants prescribed currently in the clinical practice includes the accumulation of monoamines and prolonged activation of their membrane receptors. However, a decrease in the membrane receptors density evoked by prolonged activation of monoaminergic receptors is counteracted by the second line of antidepressant activity. It is associated with the expression of inducible regulatory protein S100A10 (p11) and its partners. In this review, the authors examined the structure and function of protein p11, its interaction with such proteins as annexin A2, Ahnak, chromatinremodeling factor SMARCA3. The authors analyzed the influence of p11 on the membrane density of serotonin 5-HT1B and 5-HT4 receptors, metabotropic glutamate receptors 5, voltage-dependent potassium Kv3, and calcium Cav1.2 and 1.3 channels, that play an important role in both the effect of antidepressants and the pathogenesis of depression disorders. A systematic literature search was conducted in Scopus, Web of Science, MedLine, elibrary, and other databases.
Kuznetsov Yu.V., Evdokimov D.V., Abramets I.I. The new molecular targets for antidepressants. Medical Herald of the South of Russia. 2021;12(1):24-32. https://doi.org/10.21886/2219-8075-2021-12-1-24-32
Major depressive disorder occurs with a lifetime probability of 20% to 25% in women, 7% to 12% in men. Despite seventy years of intensive research, the pathophysiology of this disorder is yet to be fully understood. Treatment of depression is complex since the effect comes with a great latency, is not universally observed, and the therapy has multiple adverse effects [1]. Apparently, treatment of depressive disorders must be made more effective, which requires a better understanding of its pathogenesis and antidepressant pharmacology.
There are different functional classes of antidepressants today: selective and non-selective inhibitors of serotonin, noradrenaline, and dopamine reuptake, as well as monoamine oxidase inhibitors; they amplify the signaling pathway of cAMP, increase the levels of Gas in neuron membranes, and activate adenylate cyclasecoupled monoamine receptors, which activates protein kinase A (PKA) in neuron cytoplasm and nuclei. PKA, calcium/calmodulin-dependent protein kinase
type IV (CAMK4) and mitogen-activated protein kinases (MAPK) phosphorylate and activate CREB, a transcription factor that enables protein production. An increase in the activity of PKA, MAPK ERK1/2, and CREB on top of continuous administration of antidepressants results in higher levels of mRNA, neurotrophins, and growth factors. The latter enable normal neuron functional, synaptogenesis and neurogenesis in the dentate gyrus, as well as homeostatic processes in neural networks [2][3].
Therefore, the first line affected by antidepressants consists of positively adenylate cyclase-coupled monoaminergic receptors, N-methyl-D-aspartate (NMDA) glutamate receptors, metabotropic glutamate receptors, M choline receptors, etc. Since chronic administration of monoaminergic antidepressants results in a long-term increase in the extracellular concentration of monoamines in the brain, membrane receptors respond naturally to disrupted homeostasis by reducing their density. However, such chronic treatment also produces interesting collisions: the change in the membrane density of the same receptors varies from region to region in the brain. For instance, the density and activity of 5-HT1A receptors decreases in the somato-dendritic synapses of raphe nuclei while increasing in the hippocampus and in the cortex, where the levels of 5-HT1B and 5-HT4 receptors rise as well. Membrane density of alpha2 adrenoreceptors decreases in the somato-dendritic synapses of the locus coeruleus while increasing in the neurons of forebrain structures, even though the density of beta adrenoreceptors decreases there [4].
The therapeutic effects of selective serotonin reuptake inhibitors (SSRIs), which are the most popular antidepressants today, come from activating 5-HT1B and 5-HT4 receptors. However, it remains unclear why the membrane density of these receptors increases rather than decreases whilst SSRIs persistently raise the levels of serotonin in the extracellular fluid. It has been discovered in the last decade that the phenomenon is a result of serotonin receptors interacting with the inducible regulatory protein p11 also known as S100A10, nerve growth factor-induced 42C, calpactin I light chain, annexin light chain [5].
The protein is a member of S100, a family of lowmolecular-weight (11–12 kDa) acidic proteins. Its structure shown in Figure 1 has two EF hands binding Ca2+. A loop (a hinge) between the second and the third spiral enables the molecule to bend. The protein forms homodimers that can form heterotetramers with annexin A2, a cytoskeleton protein. Beside membrane receptors, p11 has been found to interact with Nav1.8, Kv, and H+ iron channels, with TRVP5 cation channels, as well as with the fragments: tissue plasminogen activator, phospholipase A2, etc. [5, 6].
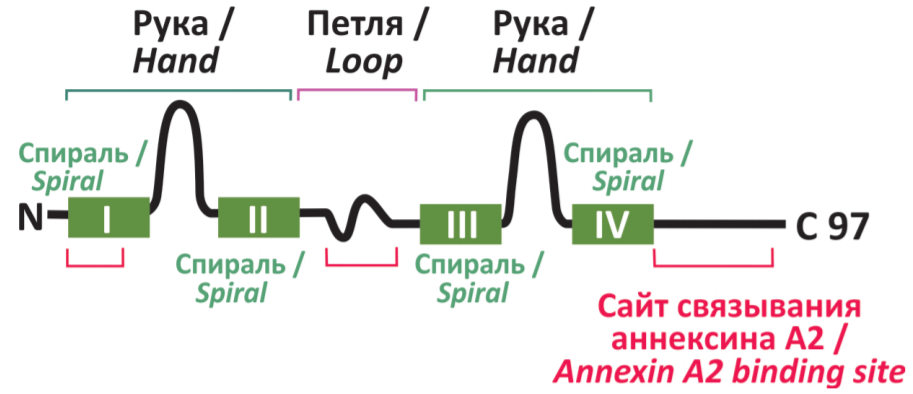
Nevertheless, the role of p11 and its targets in the development of the depressive behavioral phenotype and in antidepressant effects remains unclear due to a multitude of contradictory facts. On the one hand, administering antidepressants, levodopa (an antiParkinson medication), and serotonin (5-HT) has been found to increase the levels of mRNA and p11 in the murine cortex and hippocampus, effects mediated by the signaling pathways of neurotrophins, receptor tyrosine kinase TrkB, and protein kinase ERK. On the other hand, pro-depressive glucocorticoids, interleukins, tumor necrosis factor, and interferons also increase the levels of p11 in the rodent cortex and hippocampus [7]. Besides, non-steroidal anti-inflammatory drugs (NSAIDs) have been found to inhibit the SSRI-induced increase in p11 and related behavioral effects whilst enhancing the same effects caused by other antidepressants [8]. It is assumed that the depressogenic effect-associated increase in p11 is likely a homeostatic brain reaction that counters the development of depressive disorders.
Total p11 knockout causes the brain and the body to lose this protein, which triggers the development of depressive behavioral phenotype as shown by a change in the results of a whole behavioral test battery; it is also associated with a reduced effect of antidepressants on these indicators [9][10]. Nucleus accumbens (NAcc) has been found to be the key structure involved in the development of depressive behavior in p11-deficient rodents. Indeed, suppressing p11 production in NAcc only by administering viral mRNAi reproduces all the signs of depressive behavior like total p11 knockout does. In intact mice, p11 levels in cholinergic NAcc interneurons are 30 times those in GABAergic medium spiny neurons; removal of p11 from cholinergic interneurons induces a depressive behavioral phenotype. On the contrary, excessive virus-associated p11 production in the same interneurons but not in dorsal striatum neurons eliminates behavioral disorders in total p11 knockout mice. Interestingly, p11 deficiency in cholinergic NAcc neurons does not diminish SSRI effects in mice when immobilized in tail suspension tests [11, 12]. Therefore, NAcc p11 is required to regulate emotions and mood, but not for antidepressants to have effect.
At the same time, p11 biosynthesis in cortical neurons is necessary for antidepressants to affect rodent behavior. For instance, chronic administration of antidepressants increases cortex and hippocampus p11 levels and boosts the expression of 5-HT4 receptors in p11-containing cortical murine neurons. Removing p11 from layer 5a projection neurons inhibits the behavioral effects of SSRIs and prevents the densifying of 5-HT4 receptors in neuron membranes whilst not affecting the signs of the depressive behavioral phenotype [9][13].
Being a regulatory protein, p11 can by itself or in combination with other proteins interact with other target proteins and alter their activity. In the murine cortex, hippocampus, and NAcc, p11 is localized in close proximity to 5-HT1B/5-HT4 receptors in neuron membranes. p11 interacts with the third intracellular loop of serotonin receptors and increases their density in neuron membranes. This amplifies the cAMPmediated signaling pathways of these receptors. However, p11 gene knockout suppresses the activity of 5-HT1B/4 receptors by reducing their density in neuron membranes [14][15]. Therefore, p11 regulates membrane density rather than transcription and translation of serotonin receptors. In cultured cortical neurons, p11 enhances the effects of serotonin receptors on Ca2+ transmembrane current and protein kinase ERK1/2 activity. Serotonin and agonists of 5-HT1B receptors affect the phosphorylation of synapsin I, a presynaptic protein, and the corticostriatal glutamatergic synaptic transmission; these effects also depend on p11 [14].
Antidepressant-like effects of serotonin 1B and 4 receptor agonists in forced swim and tail suspension tests are weaker in p11 knockout mice compared to intact animals. The anxiolytic effects of these agonists are also reduced in p11-deficient mice subjected to open field tests [14]. 5-HT1B agonists have different effects on emotional learning in passive avoidance tests, disrupting it in intact mice and boosting it in p11 knockout mice. Excessive p11 expression induced by viruses in dentate gyrus and in the CA1 region of the hippocampus caused emotional learning in p11 knockout mice to recover [16].
Studies of pyramidal corticostriatal neurons of the 5a layers of motor and sensory cortex in rats found that only some of these neurons expressed p11. These neurons have a higher electrical excitability compared to p11-negative pyramidal neurons; chronic administration of SSRIs increases the density of 5-HT2A rather than 5-HT4 receptors in the former. Stressogenic effects (chronic social isolation) reduced the density of 5-HT2A receptors in p11-positive pyramidal neurons whilst chronic administration of fluoxetine reversed this effect [15].
Aside from various serotonin receptors, p11 also interacts with cG protein-dependent metabotropic glutamate receptors (mGluRs). These receptors are divided into three groups: mGluR1 and mGluR5 in Group I, mGluR2 and mGluR3 in Group II, and mGluR4/6/7/8 in Group III. Behavioral studies found mGluR5 and mGluR2/3 blockers to have a rapidly developing antidepressant effect on mice. Besides, the mGluR5 cytoplasmic tail was found to have a p11 binding site consisting of a ser-thr-val amino acids sequence. Immunoprecipitation showed that p11/ annexin A2 (Anxa2) heterotetramer was capable of binding to mGluR5. Testing on cultured HEK293 cell lines with embedded mGluR5 showed the effects of mGluR5 agonist to cause oscillations in the intracellular concentration of Ca2+, which would increase in case of excessive p11 production and decrease in case of damaged Anxa2. Therefore, p11/Anxa2 heterotetramers enhance the functioning of mGluR5. Further tests on the same cells, as well as on a COS-7 line, showed p11 and mGluR5 to co-increase their accumulation in cytoplasmic membranes, with p11 increasing the membrane density of mGluR5.
p11 and mGluR5 are found together in glutamatergic and GABAergic neurons. mGluR5 knockout in glutamatergic neurons prolonged the immobilization time in forced swim and tail suspension tests whilst reducing the sucrose preference, i.e., depressive behavioral phenotype progressed. The same procedure applied to GABAergic neurons caused an opposite change in behavior, i.e., it had an antidepressantlike effect. Similar results were obtained with p11 knocked out in GABAergic neurons. p11 knockout in glutamatergic neurons did not cause depressive behavior but made the mice more sensitive to chronic moderate stress. Another behavioral test (novelty-suppressed feeding) is used to characterize depressive and anxious behavior; this test found MPEP, an mGluR5 blocker, to have an antidepressant-like effect that would not manifest in p11 knockout mice. mGluR2/3 blocker also had an antidepressant-like effect, but that effect was not altered by removing p11. Therefore, the antidepressantlike effect of MPEP in intact mice could be eliminated by p11 knockout and reproduced by genetic deletion of mGluR5 in PV+ GABAergic interneurons [19]. The excitatory synaptic inputs of PV+ interneurons have effects that are known to stem from the activation of ionotropic AMPA and NMDA glutamate receptors, and mGluR5 increase the functional activity of NMDA receptors [20][21]. Therefore, it can be assumed that the pharmacological or genetic suppression of mGluR5 activity inhibits the activity of PV+ GABAergic inhibitory interneurons and disinhibits glutamatergic pyramidal cortical and hippocampal neurons. Behaviorwise, this manifests itself as an antidepressant-like effect.
Beside serotonin and glutamate receptors, p11 also controls the membrane density and activity of potassium channels, which determines the excitability of neurons, as well as the duration and frequency of neural spike generation. In p11 knockout mice, the hippocampus was found to have suppressed transmembrane current through Kv3.1 potassium channels that are densely packed in the membranes of parvalbumin-positive (PV+) interneurons; this suppression was associated with a lower frequency of neuron discharge in response to depolarizing the membrane with a 100 pA step in inward current. HCN cation channel currents did not change under the same circumstances. Degradation and the resulting destruction of Kv3.1 channels caused a reduction of transmembrane currents in PV+ basket cells of the dentate gyrus. Besides, p11 knockout in dentate gyrus granule cells caused mice to have double the amplitude of GABAergic IPSPs due to optogenetic stimulation of PV+ interneurons. This was due to the loss of Kv3.1 channels in axon terminals of PV+ interneurons, which limit the duration of action potentials and the presynaptic release of GABA. p11 knockout in PV+ interneurons also caused more pronounced anxious behavior in a novel environment whilst compromising their resilience to depressogenic effects of protective social stress, even though sucrose preference and immobilization time in tail suspension tests did not change [22].
PV+ interneurons are important for cognition, emotions, learning, and memory. Emotional disorders, epilepsy, or schizophrenia can develop should these neurons fail. In these neurons, Kv3 potassium channels accelerate spike repolarization and shorten the interspike interval; in presynaptic terminals of these neurons, Kv3 channels regulate GABA release and alter spike duration [23][24][25]. Interestingly, prefrontal and parietal cortical neurons have lower membrane density of Kv3.1 channels in schizophrenic patients; antipsychotics increase that density to health levels [26]. On the other hand, p11 knockout mice only had a lower density of Kv3.1 channels in hippocampus, but not in other brain structures, i.e. the mechanisms that control Kv3.1 density vary from structure to structure. Whilst chronic administration of antipsychotics increases the density of Kv3.1 channels in cortical structures, chronic administration of antidepressants reduces such density in the hippocampus. Chronically administered antidepressants have an inhibitory effect on Kv3.1 channels in PV+ dentate gyrus interneurons; this effect is associated with the activation of 5-HT5A receptors and PKA-induced phosphorylation of serine residue 503 in the Kv3.1b alpha subunit [27]. Therefore, it seems that the activity of Kv3.1 channels in PV+ interneurons is modulated by chronic administration of antidepressants, as it peaks early in treatment and is suppressed later on. Indeed, the initial effects of antidepressants cause the spike activity of PV+ interneurons to rise due to the activation of 5-HT1B receptors, a reduction in cAMP and in PKA activity, which reduces the phosphorylation of the Kv3.1 alpha subunit and boosts the functioning of Kv3.1 channels; however, the activity of these neurons attenuates further one due to down-regulation of Kv3.1 channels caused by the increased activity of 5-HT5A receptors [27].
Voltage-gated (VG) Ca2+ channels are another depression- and antidepressant-related molecular target of p11. Indeed, L type VG Ca29 channels enable the delivery of Ca2+ into neuron cytoplasm, activate CAMK4 and MAPK, and enhance the transcription processes. These channels are needed for neuron maturation, synaptic plasticity, and homeostasis of neural networks [28][29]. Genetic research has shown that Cav1.2 (CACNA1C) polymorphism is associated with a higher risk of major depressive or bipolar disorders, or schizophrenia [29].
p11 interacts with serotonin receptors and potassium channels as a heterotetramer formed with Anxa2, a cytoskeleton protein. In turn, p11/Anxa2 can form a complex with the chromatin-remodeling factor SMARCA3. Knocking out the latter does not cause depressive behavioral phenotype to emerge and develop in mice; however, it mitigates the behavioral effects of SSRIs as well as their effects on neurogenesis [30]. p11/ Anxa2 was later found to be able to form a complex with Ahnak, a high-molecular-weight (680 kDa) protein. This protein is involved in forming the gap junctions between epithelial cells in ventricular walls [30]. Ahnak was also found to be capable of binding to VG Ca2+ channel beta subunit in myocardiocytes and to be involved in regulating the density of Cav1.2 channels in the membranes of myocardiocytes, osteoblasts, and T cells [31][32].
Biochemical studies on dentate gyrus and layers 2/3 of murine prefrontal cortex found Ahnak to stabilize the p11/Anxa2 complex; besides, the N-terminus of Ahnak interacts with the pore-forming L-type Ca channel alpha subunit, whilst the C-terminus interacts with the modulatory beta subunit of the same channel and with the p11/Anxa2 complex. Testing cultured embryoderived cortical neurons revealed Ahnak knockout to reduce the total amplitude of Ca2+ currents by reducing the transmembrane current component associated with the activation of L-type calcium channels. Tests on the murine prefrontal cortex and hippocampus
sections found layer 2/3 pyramidal neurons and PV+ interneurons of the dentate gyrus in Ahnak knockout mice to have reduced amplitude of calcium currents triggered by the activation of the Cav1.2 channels. Importantly, the total number of Cav1.2 and Cav1.3 alpha subunits and additional beta subunits would not change even in light of a 50% reduction in transmembrane currents. Consequently, triple protein complex p11/ Anxa2/Ahnak regulates neuron membrane fixation of L-type calcium channels, but not the transcription or translation thereof [33].
Behavior-wise, Ahnak knockout mice had a depression-like phenotype manifesting as lower sucrose preference compared to water (a sign of anhedonia) and longer immobilization in swim and tail suspension tests. At the same time, local Ahnak knockout in glutamatergic neurons of the forebrain caused depression-like behavior in mice as indicated by a change in the behavioral test results. Local Ahnak knockout in PV+ interneurons had an antidepressant-like effect in sucrose preference and forced swim tests [33].
Interestingly, L-type Ca2+ channels regulate neuron excitability and spike generation. They employ several methods to this end: similarly to T-type Ca2+ channels (Cav3), L-type channel activation causes long-lasting depolarization of neuron membranes, which generates spikes; increased intracellular Ca2+ concentration activates BK channels, whilst depolarization excites VG Kv3.1 channels, and both types contribute to shortening the spikes while making them more frequent. This is why Ahnak knockout reduces the membrane density of L-type channels in limbic glutamatergic neurons and suppresses their activity, causing a progressive depressive behavioral phenotype. On the other hand, local Ahnak knockout in PV+ interneurons suppresses their activity as well, but it also disinhibits cortical and hippocampal pyramidal neurons as is observed during the administration of fast-acting antidepressants: ketamine and mGluR5 blockers [34][35].
In conclusion, it is safe to say the second line of antidepressant action comprises inducible regulatory proteins S100A10 (p11), annexin A2, etc., which can interact with membrane receptors and ion channels. Their intracellular carboxyl terminus or one of the intracellular loops contains a specific sequence of amino acids that determines the affinity to regulatory proteins. Besides, regulatory proteins can also interact with cytoskeleton elements. Complexes of these proteins regulate the transport of protein molecules and their fixation in membranes. Since chronic administration of antidepressants increases p11 levels in neurons, it causes a higher density of 5-HT1 and 5-HT4 receptors despite the homeostatic mechanism that has an opposite effect. On the other hand, serotoninergic and noradrenergic neurons of the afterbrain have low or no p11, causing a homeostatic response to antidepressants. That being said, complexes of p11, annexin A2, etc. stabilize and enhance the effects of monoaminergic and some fastacting (e.g. mGluR5 blockers) antidepressants. Finally, total or local p11 knockout in the brain causes depressive disorders to progress, most likely due to disrupted fixation and change in the density of receptors and ion channels in neuron membranes.
1. Belmaker RH, Agam GN. Major depressive disorder. New Engl J Med. 2008;358(1):55–68. doi: 10.1056/NEJMra073096
2. Pittenger C, Duman RC. Stress, depression, and neuroplasticity: a convergence of mechanisms. Neuropsychopharmacology. 2008;33(1):88-109. doi: 10.1038/sj.npp.1301574
3. Tiraboschi E, Tardito D, Kasahara J, Moraschi S, Pruneri P, et al. Selective phosphorylation of nuclear CREB by fluoxetine is linked to activation of CaMK IV and MAP kinase cascades. Neuropsychopharmacology. 2004;29 (10): 1831–1840. doi: 10.1038/sj.npp.1300488
4. Reuter LE, de Montigni C, Blier P. Electrophysiological characterization of the effect of long-term duloxetine administration on the rat serotonergic and noradrenergic systems. JPharmacolExpTher. 1998;285(2):404-412. PMID: 9580577
5. Svenningsson P, Greengard P. p11 (S100A10)-an inducible adaptor protein that modulates neuronal functions. Curr. Opin. Pharmacol. 2007;7(1):27–32. doi: 10.1016/j.coph.2006.10.001
6. Rescher U, Gerke V. S100A10/p11: family, friends and functions. Pflugers Arch. 2008; 455 (4):575–582. doi: 10.1007/s00424-007-0313-4
7. Svenningsson P, Kim Y, Warner-Schmidt J, Oh Y-S, Greengard P. p11 and its role in depression and therapeutic responses to antidepressants. Nat Rev Neurosci. 2013; 14(10): 673–680. doi:10.1038/nrn3564
8. Warner-Schmidt JL, Vanover KE, Chen EY, Marshall JJ, Greengard P. Antidepressant effects of selective reuptake serotonine inhibitors (SSRIs) are attenuated by anti-inflammatory drugs in mice and humans. ProcNatlAcadSci U S A. 2011;108(22):9262-9267. doi: 10.1073/pnas.1104836108
9. Warner-Schmidt JL, Chen EI, Zhang X, Marshal JJ, Morozov A, et al. A role for p11 in the antidepressant action of brain-derived neurotrophic factor. Biol. Psychiatry. 2010; 68 (6):528–535. doi: 10.1016/j.biopsych.2010.04.029
10. Egeland M, Warner-Schmidt J, Greengard P, Svenningsson P. Neurogenic effects of fluoxetine are attenuated in p11 (S100A10) knockout mice. Biol. Psychiatry. 2010; 67 (11):1048–1056. doi: 10.1016/j.biopsych.2010.01.024
11. Warner-Schmidt JL, Schmidt EF, Marshall JJ, Rubin AJ, Arango-Lievano M, et al. Cholinergic interneurons in the nucleus accumbens regulate depression-like behavior. Proc. Natl Acad. Sci. USA. 2012; 109 (28):11360–11365. doi: 10.1073/pnas.1209293109
12. Alexander B, Warner-Schmidt JL, Eriksson TM, Tamminga C, Arango-Lievano M, et al. Reversal of depressed behaviors in mice by p11 gene therapy in the nucleus accumbens. Science Transl Med. 2010; 2 (54):54ra76. doi: 10.1126/scitranslmed.3001079
13. Schmidt EF, Warner-Schmidt JL, Otopalik BG, Pickett SB, Greengard P, Heintz N. Identification of the cortical neurons that mediate antidepressant responses. Cell. 2012; 149 (5):1152–1163. doi: 10.1016/j.cell.2012.03.038
14. Svenningsson P, Chergui K, Rachleff I, Flajolet M, Zhang XEl, et al. Alterations in 5-HT1B receptor function by p11 in depression-like states. Science. 2006; 311 (5757): 77–80. doi:10.1126/science.1117571
15. Warner-Schmidt JL, Flajolet M, Maller A, Chen EY, Qi H, et al. Role of p11 in cellular and behavioral effects of 5-HT4 receptorstimulation. J. Neurosci. 2009; 29 (6): 1937–1946. doi:10.1523/JNEUROSCI.5343-08.2009
16. Eriksson TM, Delagrange P, Spedding M, Popoli M, Mate AA, et al. Bidirectional regulation of emotional memory by 5-HT1B receptors involves hippocampal p11. Mol. Psychiatry. 2012; 17 (2): 173-184. doi10.1038/mp.2010.131
17. Sargin D, Chottekalapanda RU, Perit KE, Yao V, Chu D, et al. Mapping the physiological and molecular markers of stress and SSRI antidepressant treatment in S100a10 corticostriatal neurons. Molecular Psychiatry. 2020; 25 (6):1112–1129. doi: 10.1038/s41380-019-0473-6
18. Conn PJ, Pin JP. Pharmacology and functions of metabotropic glutamate receptors. Annu Rev PharmacolToxicol. 1997; 37:205–237. doi: 10.1146/annurev.pharmtox.37.1.205
19. Lee K-W, Westin L, Kim J, Chang JC, Oh Y-S, et al. Alteration by p11 of mGluR5 localization regulates depressionlike behaviors. Mol Psychiatry. 2015; 20(12): 1546–1556. doi:10.1038/mp.2015.132
20. Awad H, Hubert GW, Smith Y, Levey AI, Conn PJ. Activation of metabotropic glutamate receptor 5 has direct excitatory effects and potentiates NMDA receptor currents in neurons of the subthalamic nucleus. J Neurosci. 2000; 20(21):7871–7879. doi: 10.1523/JNEUROSCI.20-21-07871.2000
21. Rosenbrock H, Kramer G, Hobson S, Koros E, Grundl M, et al. Functional interaction of metabotropic glutamate receptor 5 and NMDA-receptor by a metabotropic glutamate receptor 5 positive allosteric modulator. Eur J Pharmacol. 2010; 639(1–3):40–46. doi: 10.1016/j.ejphar.2010.02.057
22. Seo SJ, Sveninngsson P. Modulation of ion channels and receptors by p11 (S100A10). Trends Pharmacol Sci. 2020; 41 (7): 487-497. doi: 10.1016/j.tips.2020.04.004
23. Rudy B, McBain CJ. Kv3 channels: voltage-gated K1 channels designed for high-frequency repetitive firing. Trends Neurosci. 2001; 24:517–526. doi:10.1016/s0166-2236(00)01892-0
24. Hoppa MB, Gouzer G, Armbruster M, Ryan TA. Control andplasticity of the presynaptic action potential waveform at small CNS nerve terminals. Neuron. 2014; 84 (4):778–789. doi: 10.1016/j.neuron.2014.09.038
25. Hu H, Roth FC, Vandael D, Jonas P. Complementary tuning of Na+ and K+ channel gating underlies fast and energy-efficient actionpotentials in GABAergic interneuron axons. Neuron. 2018;98(1):156–165.E6. doi: 10.1016/j.neuron.2018.02.024
26. Yanagi M, Joho RH, Southcott SA, Shukla AA, Ghose S, Tamminga CA. Kv3.1-containing K(1) channels are reduced in untreated schizophrenia and normalized with antipsychotic drugs. MolPsychiatry. 2014; 19 (5):573–579. doi: 10.1038/mp.2013.49
27. Sagi Y, Medrihan L, George K, Barney M, McCabe KA, Greengard P. Emergence of 5-HT5A signaling in parvalbumin neurons mediates delayed antidepressant action. MolPsychiatry. 2020;25 (6):1191–1201. doi: 10.1038/mp.2013.49
28. Simms BA, Zamponi GW. Neuronal voltage-gated calcium channels: structure, function, and dysfunction. Neuron. 2014;82 (1):24–45. doi: 10.1016/j.neuron.2014.03.016
29. Heyes S, Pratt WS, Rees E, Dahimene S, Ferron L, et al. Genetic disruption of voltage-gated calcium channels in psychiatric and neurological disorders. ProgNeurobiol. 2015;134 (Nov):36–54. doi: 10.1016/j.pneurobio.2015.09.002
30. Oh YS, Gao P, Lee KW, Ceglia I, Seo JS, et al. SMARCA3, a chromatin-remodeling factor, is required for p11-dependent antidepressant action. Cell. 2013;152 (4):831–843. doi: 10.1016/j.cell.2013.01.014
31. Alvarez J, Hamplova J, Hohaus A, Morano I, Haase H, Vassort G. Calcium current in rat cardiomyocytes is modulated by the carboxylterminal ahnak domain. J Biol Chem. 2004;279 (13):12456–12461. doi: 10.1074/jbc.M31217720
32. Matza D, Badou A, Kobayashi KS, Goldsmith-Pestana K, Masuda Y, et al. A scaffold protein, AHNAK1, is required for calcium signaling during T cell activation. Immunity. 2008;28 (1):64–74. doi: 10.1016/j.immuni.2007.11.020
33. Jin J, Bhatti DL, Lee K-W, Medrihan L, Jia Cheng J, et al. Ahnak scaffolds p11/Anxa2 complex and L-type voltage-gatedcalcium channel and modulates depressive behavior. Mol Psychiatry. 2020; 25 (5):1035–1049. doi:10.1038/s41380-019-0371-y
34. Striessnig J, Pinggera A, Kaur G, Bock G, Tuluc P. L-type Ca(2+) channels in heart and brain. Wiley Interdiscip Rev MembrTranspSignal. 2014;3 (2):15–38. doi: 10.1002/wmts.102
35. Liu Y, Harding M, Pittman A, Dore J, Striessnig J, et al. Cav1.2 and Cav1.3 L-type calcium channels regulate dopaminergic firing activity in the mouse ventral tegmental area. J Neurophysiol. 2014;112 (5):1119–1130. doi:10.1152/jn.00757.2013
Yuriy V. Kuznetsov, Cand. Sci. (Med), associate Professor of the I.V. Komissarov Department of Pharmacology and Clinical Pharmacology
Donetsk, DPR
Dmitriy V. Evdokimov, Cand. Sci. (Med), associate Professor of the I.V. Komissarov Department of Pharmacology and Clinical Pharmacology
Donetsk, DPR
Igor I. Abramets. Dr. Sci. (Med), full Professor of the I.V. Komissarov Department of Pharmacology and Clinical Pharmacology
Donetsk, DPR
Kuznetsov Yu.V., Evdokimov D.V., Abramets I.I. The new molecular targets for antidepressants. Medical Herald of the South of Russia. 2021;12(1):24-32. https://doi.org/10.21886/2219-8075-2021-12-1-24-32
29, Nakhichevansky Lane, Rostov-on-Don, 344002
Rostov State Medical University
Тel.: +79185710558
e-mail: journal@medicalherald.ru


































